Citation: Yun Kang, Sourav Kumar Sasmal, Komi Messan. A two-patch prey-predator model with predator dispersal driven by the predation strength[J]. Mathematical Biosciences and Engineering, 2017, 14(4): 843-880. doi: 10.3934/mbe.2017046

| [1] | [ L. Aarssen,R. Turkington, Biotic specialization between neighbouring genotypes in lolium perenne and trifolium repens from a permanent pasture, The Journal of Ecology, 73 (1985): 605-614. |
| [2] | [ R. F. Alder, Migration alone can produce persistence of host-parasitoid models, The American Naturalist, 141 (1993): 642-650. |
| [3] | [ J. Bascompte,R. V. Solé, Spatially induced bifurcations in single-species population dynamics, Journal of Animal Ecology, 63 (1994): 256-264. |
| [4] | [ B. M. Bolker,S. W. Pacala, Spatial moment equations for plant competition: Understanding spatial strategies and the advantages of short dispersal, The American Naturalist, 153 (1999): 575-602. |
| [5] | [ C. J. Bolter,M. Dicke,J. J. Van Loon,J. Visser,M. A. Posthumus, Attraction of colorado potato beetle to herbivore-damaged plants during herbivory and after its termination, Journal of Chemical Ecology, 23 (1997): 1003-1023. |
| [6] | [ C. Carroll,D. H. Janzen, Ecology of foraging by ants, Annual Review of Ecology and Systematics, 4 (1973): 231-257. |
| [7] | [ A. Casal,J. Eilbeck,J. López-Gómez, Existence and uniqueness of coexistence states for a predator-prey model with diffusion, Differential and Integral Equations, 7 (1994): 411-439. |
| [8] | [ P. L. Chesson,W. W. Murdoch, Aggregation of risk: Relationships among host-parasitoid models, American Naturalist, 127 (1986): 696-715. |
| [9] | [ W. C. Chewning, Migratory effects in predator-prey models, Mathematical Biosciences, 23 (1975): 253-262. |
| [10] | [ R. Cressman,K. Vlastimil, Two-patch population models with adaptive dispersal: The effects of varying dispersal speeds, Journal of Mathematical Biology, 67 (2013): 329-358. |
| [11] | [ E. Curio, The Ethology of Predation ,Springer-Verlag Berlin Heidelberg, 7 1976. |
| [12] | [ M. Doebli, Dispersal and dynamics, Theoretical Population Biology, 47 (1995): 82-106. |
| [13] | [ W. Feng,B. Rock,J. Hinson, On a new model of two-patch predator-prey system with migration of both species, Journal of Applied Analysis and Computation, 1 (2011): 193-203. |
| [14] | [ J. Ford, The Role of the Trypanosomiases in African Ecology. A Study of the Tsetse Fly Problem, in Oxford University Press, Oxford, 1971. |
| [15] | [ A. G. Gatehouse, Permanence and the dynamics of biological systems, Host Finding Behaviour Of Tsetse Flies, null (1972): 83-95. |
| [16] | [ S. Ghosh,S. Bhattacharyya, A two-patch prey-predator model with food-gathering activity, Journal of Applied Mathematics and Computing, 37 (2011): 497-521. |
| [17] | [ M. Gillies,T. Wilkes, The range of attraction of single baits for some West African mosquitoes, Bulletin of Entomological Research, 60 (1970): 225-235. |
| [18] | [ M. Gillies,T. Wilkes, The range of attraction of animal baits and carbon dioxide for mosquitoes, Bulletin of Entomological Research, 61 (1972): 389-404. |
| [19] | [ M. Gillies,T. Wilkes, The range of attraction of birds as baits for some west african mosquitoes (diptera, culicidae), Bulletin of Entomological Research, 63 (1974): 573-582. |
| [20] | [ I. Hanski, null, Metapopulation Ecology, Oxford University Press, Oxford, 1999. |
| [21] | [ I. A. Hanski,M. E. Gilpin, null, Metapopulation Biology: Ecology, Genetics, and Evolution, Academic Press, San Diego, 1997. |
| [22] | [ M. Hassell,R. May, Aggregation of predators and insect parasites and its effect on stability, The Journal of Animal Ecology, 43 (1974): 567-594. |
| [23] | [ M. Hassell,T. Southwood, Foraging strategies of insects, Annual Review of Ecology and Systematics, 9 (1978): 75-98. |
| [24] | [ M. Hassell,O. Miramontes,P. Rohani,R. May, Appropriate formulations for dispersal in spatially structured models: comments on bascompte & Solé, Journal of Animal Ecology, 64 (1995): 662-664. |
| [25] | [ M. P. Hassell,H. N. Comins,R. M. May, Spatial structure and chaos in insect population dynamics, Nature, 353 (1991): 255-258. |
| [26] | [ A. Hastings, Can spatial variation along lead to selection for dispersal?, Theoretical Population Biology, 24 (1983): 244-251. |
| [27] | [ A. Hastings, Complex interactions between dispersal and dynamics: Lessons from coupled logistic equations, Ecology, 74 (1993): 1362-1372. |
| [28] | [ C. Hauzy,M. Gauduchon,F. D. Hulot,M. Loreau, Density-dependent dispersal and relative dispersal affect the stability of predator-prey metacommunities, Journal of Theoretical Biology, 266 (2010): 458-469. |
| [29] | [ R. D. Holt, Population dynamics in two-patch environments: Some anomalous consequences of an optimal habitat distribution, Theoretical Population Biology, 28 (1985): 181-208. |
| [30] | [ S. Hsu,S. Hubbell,P. Waltman, A mathematical theory for single-nutrient competition in continuous cultures of micro-organisms, SIAM Journal on Applied Mathematics, 32 (1977): 366-383. |
| [31] | [ S. Hsu, On global stability of a predator-prey system, Mathematical Biosciences, 39 (1978): 1-10. |
| [32] | [ Y. Huang,O. Diekmann, Predator migration in response to prey density: What are the consequences?, Journal of Mathematical Biology, 43 (2001): 561-581. |
| [33] | [ V. Hutson, A theorem on average liapunov functions, Monatshefte für Mathematik, 98 (1984): 267-275. |
| [34] | [ V. Hutson,K. Schmit, Permanence and the dynamics of biological systems, Mathematical Biosciences, 111 (1992): 1-71. |
| [35] | [ V. A. Jansen, Regulation of predator-prey systems through spatial interactions: A possible solution to the paradox of enrichment, Oikos, 74 (1995): 384-390. |
| [36] | [ V. A. Jansen, The dynamics of two diffusively coupled predator-prey populations, Theoretical Population Biology, 59 (2001): 119-131. |
| [37] | [ V. A. A. Jansen, Theoretical Aspects of Metapopulation Dynamics, PhD thesis, Ph. D. thesis, Leiden University, The Netherlands, 1994. |
| [38] | [ Y. Kang,D. Armbruster, Dispersal effects on a discrete two-patch model for plant-insect interactions, Journal of Theoretical Biology, 268 (2011): 84-97. |
| [39] | [ Y. Kang,C. Castillo-Chavez, Multiscale analysis of compartment models with dispersal, Journal of Biological Dynamics, 6 (2012): 50-79. |
| [40] | [ P. Kareiva,G. Odell, Swarms of predators exhibit "prey-taxis" if individual predators use area-restricted search, American Naturalist,, 130 (1987): 233-270. |
| [41] | [ P. Kareiva,A. Mullen,R. Southwood, Population dynamics in spatially complex environments: Theory and data [and discussion], Philosophical Transactions of the Royal Society of London B: Biological Sciences, 330 (1990): 175-190. |
| [42] | [ S. Kéfi,M. Rietkerk,M. van Baalen,M. Loreau, Local facilitation, bistability and transitions in arid ecosystems, Theoretical Population Biology, 71 (2007): 367-379. |
| [43] | [ P. Klepac,M. G. Neubert,P. van den Driessche, Dispersal delays, predator-prey stability, and the paradox of enrichment, Theoretical Population Biology, 71 (2007): 436-444. |
| [44] | [ M. Kummel,D. Brown,A. Bruder, How the aphids got their spots: Predation drives self-organization of aphid colonies in a patchy habitat, Oikos, 122 (2013): 896-906. |
| [45] | [ K. Kuto,Y. Yamada, Multiple coexistence states for a prey-predator system with cross-diffusion, Journal of Differential Equations, 197 (2004): 315-348. |
| [46] | [ I. Lengyel,I. R. Epstein, Diffusion-induced instability in chemically reacting systems: Steady-state multiplicity, oscillation, and chaos, Chaos: An Interdisciplinary Journal of Nonlinear Science, 1 (1991): 69-76. |
| [47] | [ S. A. Levin, Dispersion and population interactions, American Naturalist, 108 (1974): 207-228. |
| [48] | [ R. Levins, Some demographic and genetic consequences of environmental heterogeneity for biological control, Bulletin of the Entomological Society of America, 15 (1969): 237-240. |
| [49] | [ Z.-z. Li,M. Gao,C. Hui,X.-z. Han,H. Shi, Impact of predator pursuit and prey evasion on synchrony and spatial patterns in metapopulation, Ecological Modelling, 185 (2005): 245-254. |
| [50] | [ X. Liu,L. Chen, Complex dynamics of Holling type Ⅱ Lotka--Volterra predator--prey system with impulsive perturbations on the predator, Chaos, Solitons & Fractals, 16 (2003): 311-320. |
| [51] | [ Y. Liu, The Dynamical Behavior of a Two Patch Predator-Prey Model ,Honor Thesis, from The College of William and Mary, 2010. |
| [52] | [ J. H. Loughrin,D. A. Potter,T. R. Hamilton-Kemp,M. E. Byers, Role of feeding-induced plant volatiles in aggregative behavior of the japanese beetle (coleoptera: Scarabaeidae), Environmental Entomology, 25 (1996): 1188-1191. |
| [53] | [ J. Madden, Physiological reactions of Pinus radiata to attack by woodwasp, Sirex noctilio F.(Hymenoptera: Siricidae), Bulletin of Entomological Research, 67 (1977): 405-426. |
| [54] | [ L. Markus, Ⅱ. Asymptotically autonomous differential systems, in Contributions to the Theory of Nonlinear Oscillations (AM-36), Vol. Ⅲ, Princeton University Press, 1956, 17–30. |
| [55] | [ R. M. May, Host-parasitoid systems in patchy environments: A phenomenological model, The Journal of Animal Ecology, 47 (1978): 833-844. |
| [56] | [ R. McMurtrie, Persistence and stability of single-species and prey-predator systems in spatially heterogeneous environments, Mathematical Biosciences, 39 (1978): 11-51. |
| [57] | [ T. F. Miller,D. J. Mladenoff,M. K. Clayton, Old-growth northern hardwood forests: Spatial autocorrelation and patterns of understory vegetation, Ecological Monographs, 72 (2002): 487-503. |
| [58] | [ W. W. Murdoch,C. J. Briggs,R. M. Nisbet,W. S. Gurney,A. Stewart-Oaten, Aggregation and stability in metapopulation models, American Naturalist, 140 (1992): 41-58. |
| [59] | [ M. Pascual, Diffusion-induced chaos in a spatial predator-prey system, Proceedings of the Royal Society of London B: Biological Sciences, 251 (1993): 1-7. |
| [60] | [ M. Rees,P. J. Grubb,D. Kelly, Quantifying the impact of competition and spatial heterogeneity on the structure and dynamics of a four-species guild of winter annuals, American Naturalist, 147 (1996): 1-32. |
| [61] | [ M. Rietkerk,J. Van de Koppel, Regular pattern formation in real ecosystems, Trends in Ecology & Evolution, 23 (2008): 169-175. |
| [62] | [ P. Rohani,G. D. Ruxton, Dispersal and stability in metapopulations, Mathematical Medicine and Biology, 16 (1999): 297-306. |
| [63] | [ M. L. Rosenzweig,R. H. MacArthur, Graphical representation and stability conditions of predator-prey interactions, American Naturalist, 97 (1963): 209-223. |
| [64] | [ G. D. Ruxton, Density-dependent migration and stability in a system of linked populations, Bulletin of Mathematical Biology, 58 (1996): 643-660. |
| [65] | [ L. M. Schoonhoven, Plant recognition by lepidopterous larvae, (1972), 87–99. |
| [66] | [ L. M. Schoonhoven, On the variability of chemosensory information, The Host-Plant in Relation to Insect Behaviour and Reproduction, Symp. Biol. Hung., 16 (1976), 261–266. |
| [67] | [ L. M. Schoonhoven, Chemosensory systems and feeding behavior in phytophagous insects, (1977), 391–398. |
| [68] | [ E. W. Seabloom,O. N. Bjørnstad,B. M. Bolker,O. Reichman, Spatial signature of environmental heterogeneity, dispersal, and competition in successional grasslands, Ecological Monographs, 75 (2005): 199-214. |
| [69] | [ G. Seifert and L. Markus, Contributions to the Theory of Nonlinear Oscillations ,Princeton University Press, 1956. |
| [70] | [ Y. Shahak,E. Gal,Y. Offir,D. Ben-Yakir, Photoselective shade netting integrated with greenhouse technologies for improved performance of vegetable and ornamental crops, International Workshop on Greenhouse Environmental Control and Crop Production in Semi-Arid Regions, 797 (2008): 75-80. |
| [71] | [ R. V. Solé and J. Bascompte, Self-Organization in Complex Ecosystems ,Princeton University Press, Princeton, 2006. |
| [72] | [ A. Soro,S. Sundberg,H. Rydin, Species diversity, niche metrics and species associations in harvested and undisturbed bogs, Journal of Vegetation Science, 10 (1999): 549-560. |
| [73] | [ H. R. Thieme, Mathematics in Population Biology ,Princeton University Press, 2003. |
| [74] | [ D. Tilman and P. M. Kareiva, Spatial Ecology: The Role of Space in Population Dynamics and Interspecific Interactions ,volume 30, Princeton University Press, 1997. |
| [75] | [ J. van de Koppel,J. C. Gascoigne,G. Theraulaz,M. Rietkerk,W. M. Mooij,P. M. Herman, Experimental evidence for spatial self-organization and its emergent effects in mussel bed ecosystems, Science, 322 (2008): 739-742. |
| [76] | [ J. K. Waage, Behavioral Aspects of Foraging in the Parasitoid, Nemeritis Canescens (Grav. ) ,PhD Thesis, from University of London, 1977. |
| [77] | [ J. Wang,J. Shi,J. Wei, Predator-prey system with strong Allee effect in prey, Journal of Mathematical Biology, 62 (2011): 291-331. |

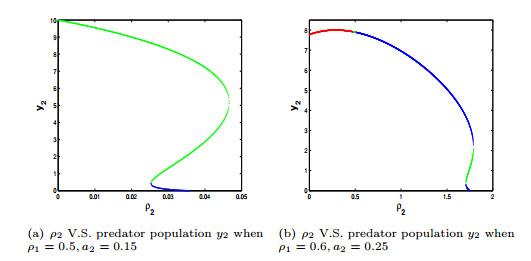

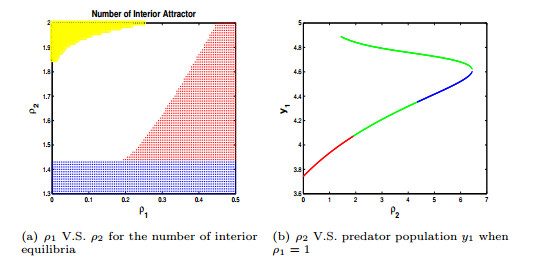
Figures(4) / Tables(3)

Yun Kang, Sourav Kumar Sasmal, Komi Messan. A two-patch prey-predator model with predator dispersal driven by the predation strength[J]. Mathematical Biosciences and Engineering, 2017, 14(4): 843-880. doi: 10.3934/mbe.2017046







 DownLoad:
DownLoad: