Citation: Somisetti V. Sambasivarao, David M. Granum, Hua Wang, C. Mark Maupin. Identifying the Enzymatic Mode of Action for Cellulase Enzymes by Means of Docking Calculations and a Machine Learning Algorithm[J]. AIMS Molecular Science, 2014, 1(1): 59-80. doi: 10.3934/molsci.2014.1.59

| [1] |
Bhat MK, Bhat S (1997) Cellulose degrading enzymes and their potential industrial applications. Biotechnol Advances 15: 583-620. doi: 10.1016/S0734-9750(97)00006-2

|
| [2] |
Himmel ME, Ding SY, Johnson DK, et al. (2007) Biomass recalcitrance: Engineering plants and enzymes for biofuels production. Science 315: 804-807. doi: 10.1126/science.1137016
|
| [3] | Himmel ME (2008) Biomass Recalcitrance: Deconstructing the Plant Cell Wall for Bioenergy. Wiley Blackwell. |
| [4] | Updegraff DM (1969) Semimicro determination of cellulose in biological materials. Anal Biochem 420-424. |
| [5] |
Wang LS, Zhang YZ, Gao PJ (2008) A novel function for the cellulose binding module of cellobiohydrolase I. Sci Chin Series C-Life Sci 51: 620-629. doi: 10.1007/s11427-008-0088-3
|
| [6] |
Edgar KJ, Buchanan CM, Debenham JS, et al. (2001) Advances in cellulose ester performance and application. Prog Polym Sci 26: 1605-1688. doi: 10.1016/S0079-6700(01)00027-2
|
| [7] |
Ragauskas AJ, Williams CK, Davison BH, et al. (2006) The path forward for biofuels and biomaterials. Science 311: 484-489. doi: 10.1126/science.1114736
|
| [8] |
Lynd LR, Laser MS, Brandsby D, et al. (2008) How biotech can transform biofuels. Nat Biotechnol 26: 169-172. doi: 10.1038/nbt0208-169
|
| [9] |
Schubert C (2006) Can biofuels finally take center stage? Nat Biotechnol 24: 777-784. doi: 10.1038/nbt0706-777
|
| [10] |
Andre G, Kanchanawong P, Palma R, et al. (2003) Computational and experimental studies of the catalytic mechanism of Thermobifida fusca cellulase Cel6A (E2). Protein Eng 16: 125-134. doi: 10.1093/proeng/gzg017
|
| [11] |
Wolfenden R, Yuan Y (2008) Rates of spontaneous cleavage of glucose, fructose, sucrose, and trehalose in water, and the catalytic proficiencies of invertase and trehalas. J Am Chem Soc 130: 7548. doi: 10.1021/ja802206s
|
| [12] |
Nishiyama Y, Sugiyama J, Chanzy H, et al. (2003) Crystal structure and hydrogen bonding system in cellulose 1(alpha), from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 125: 14300-14306. doi: 10.1021/ja037055w
|
| [13] |
CarleUrioste JC, EscobarVera J, ElGogary S, et al. (1997) Cellulase induction in Trichoderma reesei by cellulose requires its own basal expression. J Biol Chem 272: 10169-10174. doi: 10.1074/jbc.272.15.10169
|
| [14] |
Bayer EA, Chanzy H, Lamed R, et al. (1998) Cellulose, cellulases and cellulosomes. Curr Opin Structl Biol 8: 548-557. doi: 10.1016/S0959-440X(98)80143-7
|
| [15] |
Boisset C, Fraschini C, Schulein M, et al. (2000) Imaging the enzymatic digestion of bacterial cellulose ribbons reveals the endo character of the cellobiohydrolase Cel6A from Humicola insolens and its mode of synergy with cellobiohydrolase Cel7A. Appl Environ Microbiol 66: 1444-1452. doi: 10.1128/AEM.66.4.1444-1452.2000
|
| [16] |
Cantarel BL, Coutinho PM, Rancurel C, et al. (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37: D233-D238. doi: 10.1093/nar/gkn663
|
| [17] |
Wilson DB (2009) Cellulases and biofuels. Curr Opin Biotechnol 20: 295-299. doi: 10.1016/j.copbio.2009.05.007
|
| [18] |
Davies G, Henrissat B (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3: 853-859. doi: 10.1016/S0969-2126(01)00220-9
|
| [19] |
Henrissat B, Davies G (1997) Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol 7: 637-644. doi: 10.1016/S0959-440X(97)80072-3
|
| [20] |
Divne C, Stahlberg J, Teeri TT, et al. (1998) High-resolution crystal structures reveal how a cellulose chain is bound in the 50 angstrom long tunnel of cellobiohydrolase I from Trichoderma reesei. J Mol Biol 275: 309-325. doi: 10.1006/jmbi.1997.1437
|
| [21] | Davies GJ, Brzozowski AM, Dauter M, et al. (2000) Structure and function of Humicola insolens family 6 cellulases: structure of the endoglucanase, Cel6B, at 1.6 angstrom resolution. Biochem J 348: 201-207. |
| [22] |
Yang B, Willies DM, Wyman CE (2006) Changes in the enzymatic hydrolysis rate of avicel cellulose with conversion. Biotechnol Bioeng 94: 1122-1128. doi: 10.1002/bit.20942
|
| [23] |
Dashtban M, Maki M, Leung KT, et al. (2010) Cellulase activities in biomass conversion: measurement methods and comparison. Crit Rev Biotechnol 30: 302-309. doi: 10.3109/07388551.2010.490938
|
| [24] |
Teeri TT (1997) Crystalline cellulose degradation: New insight into the function of cellobiohydrolases. Trends Biotechnol 15: 160-167. doi: 10.1016/S0167-7799(97)01032-9
|
| [25] |
Rouvinen J, Bergfors T, Teeri T, et al. (1990) 3-dimensional structure of cellobiohydrolase-II from trichoderma-reesei. Science 249: 380-386. doi: 10.1126/science.2377893
|
| [26] |
Meinke A, Damude HG, Tomme P, et al. (1995) Enhancement of the endo-beta-1,4-glucanase activity of an exocellobiohydrolase by deletion of a surface loop. J Biol Chem 270: 4383-4386. doi: 10.1074/jbc.270.9.4383
|
| [27] |
Kurasin M, Valjamae P (2011) Processivity of Cellobiohydrolases Is Limited by the Substrate. J Biol Chem 286: 169-177. doi: 10.1074/jbc.M110.161059
|
| [28] |
Breyer WA, Matthews BW (2001) A structural basis for processivity. Protein Sci 10: 1699-1711. doi: 10.1110/ps.10301
|
| [29] |
Morris GM, Huey R, Lindstrom W, et al. (2009) AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem 30: 2785-2791. doi: 10.1002/jcc.21256
|
| [30] |
Pettersen EF, Goddard TD, Huang CC, et al. (2004) UCSF chimera - A visualization system for exploratory research and analysis. J Comput Chem 25: 1605-1612. doi: 10.1002/jcc.20084
|
| [31] |
Wang H, Nie F, Huang H, et al. (2012) Identifying quantitative trait loci via group-sparse multitask regression and feature selection: an imaging genetics study of the ADNI cohort. Bioinformatics 28: 229-237. doi: 10.1093/bioinformatics/btr649
|
| [32] |
Wang H, Nie F, Huang H, et al. (2012) Identifying disease sensitive and quantitative trait-relevant biomarkers from multidimensional heterogeneous imaging genetics data via sparse multimodal multitask learning. Bioinformatics 28: i127-136. doi: 10.1093/bioinformatics/bts228
|
| [33] | Lee S, Zhu J, Xing EP (2010) Adaptive Multi-Task Lasso: with application to eQTL detection. Adv Neural Informat Process Syst: 1306-1314. |
| [34] |
Puniyani K, Kim S, Xing EP (2010) Multi-population GWA mapping via multi-task regularized regression. Bioinformatics 26: i208-216. doi: 10.1093/bioinformatics/btq191
|
| [35] | Mackenzie LF, Sulzenbacher G, Divne C, et al. (1998) Crystal structure of the family 7 endoglucanase I (Cel7B) from Humicola insolens at 2.2 angstrom resolution and identification of the catalytic nucleophile by trapping of the covalent glycosyl-enzyme intermediate. Biochem J 335: 409-416. |
| [36] |
Zou JY, Kleywegt GJ, Stahlberg J, et al. (1999) Crystallographic evidence for substrate ring distortion and protein conformational changes during catalysis in cellobiohydrolase Cel6A from Trichoderma reesei. Struct Fold Des 7: 1035-1045. doi: 10.1016/S0969-2126(99)80171-3
|
| [37] |
Parkkinen T, Koivula A, Vehmaanpera J, et al. (2008) Crystal structures of Melanocarpus albomyces cellobiohydrolase Ce17B in complex with cello-oligomers show high flexibility in the substrate binding. Protein Sci 17: 1383-1394. doi: 10.1110/ps.034488.108
|
| [38] |
Varrot A, Schulein M, Davies GJ (2000) Insights into ligand-induced conformational change in Cel5A from Bacillus agaradhaerens revealed by a catalytically active crystal form. J Mol Biol 297: 819-828. doi: 10.1006/jmbi.2000.3567
|
| [39] |
Divne C, Stahlberg J, Reinikainen T, et al. (1994) The 3-dimensional crystal-structure of the catalytic core of cellobiohydroase-I from trichoderma-reesei. Science 265: 524-528. doi: 10.1126/science.8036495
|
| [40] |
Grassick A, Murray PG, Thompson R, et al. (2004) Three-dimensional structure of a thermostable native cellobiohydrolase, CBHIB, and molecular characterization of the cel7 gene from the filamentous fungus, Talaromyces emersonii. Eur J Biochem 271: 4495-4506. doi: 10.1111/j.1432-1033.2004.04409.x
|
| [41] |
Momeni MH, Payne CM, Hansson H, et al. (2013) Structural, Biochemical, and Computational Characterization of the Glycoside Hydrolase Family 7 Cellobiohydrolase of the Tree-killing Fungus Heterobasidion irregulare. J Biol Chem 288: 5861-5872. doi: 10.1074/jbc.M112.440891
|
| [42] | Kleywegt GJ, Zou JY, Divne C, et al. (1997) The crystal structure of the catalytic core domain of endoglucanase I from Trichoderma reesei at 3.6 angstrom resolution, and a comparison with related enzymes. J Mol Biol 272: 383-397. |
| [43] | Munoz IG, Ubhayasekera W, Henriksson H, et al. (2001) Family 7 cellobiohydrolases from Phanerochaete chrysosporium: Crystal structure of the catalytic module of Cel7D (CBH58) at 1.32 angstrom resolution and homology models of the isozymes. J Mol Biol 314: 1097-1111. |
| [44] |
Ubhayasekera W, Munoz IG, Vasella A, et al. (2005) Structures of Phanerochaete chrysosporium Cel7D in complex with product and inhibitors. Febs J 272: 1952-1964. doi: 10.1111/j.1742-4658.2005.04625.x
|
| [45] | Varrot A, Hastrup S, Schulein M, et al. (1999) Crystal structure of the catalytic core domain of the family 6 cellobiohydrolase II, Cel6A, from Humicola insolens, at 1.92 angstrom resolution. Biochem J 337: 297-304. |
| [46] |
Varrot A, Frandsen TP, von Ossowski I, et al. (2003) Structural basis for ligand binding and processivity in cellobiohydrolase Cel6A from Humicola insolens. Structure 11: 855-864. doi: 10.1016/S0969-2126(03)00124-2
|
| [47] |
Larsson AM, Bergfors T, Dultz E, et al. (2005) Crystal structure of Thermobifida fusca endoglucanase Cel6A in complex with substrate and inhibitor: The role of tyrosine Y73 in substrate ring distortion. Biochemistry 44: 12915-12922. doi: 10.1021/bi0506730
|
| [48] |
Varrot A, Schulein M, Davies GJ (1999) Structural changes of the active site tunnel of Humicola insolens cellobiohydrolase, Cel6A, upon oligosaccharide binding. Biochemistry 38: 8884-8891. doi: 10.1021/bi9903998
|
| [49] |
Liu Y, Yoshida M, Kurakata Y, et al. (2010) Crystal structure of a glycoside hydrolase family 6 enzyme, CcCel6C, a cellulase constitutively produced by Coprinopsis cinerea. Febs J 277: 1532-1542. doi: 10.1111/j.1742-4658.2010.07582.x
|
| [50] | Davies GJ, Dauter M, Brzozowski AM, et al. (1998) Structure of the Bacillus agaradherans family 5 endoglucanase at 1.6 angstrom and its cellobiose complex at 2.0 angstrom resolution. Biochemistry 37: 1926-1932. |
| [51] |
Wu TH, Huang CH, Ko TP, et al. (2011) Diverse substrate recognition mechanism revealed by Thermotoga maritima Cel5A structures in complex with cellotetraose, cellobiose and mannotriose. Biochim Et Biophys Acta-Proteins and Proteomics 1814: 1832-1840. doi: 10.1016/j.bbapap.2011.07.020
|
| [52] |
Lee TM, Farrow MF, Arnold FH, et al. (2011) A structural study of Hypocrea jecorina Cel5A. Protein Sci 20: 1935-1940. doi: 10.1002/pro.730
|
| [53] |
Pereira JH, Sapra R, Volponi JV, et al. (2009) Structure of endoglucanase Cel9A from the thermoacidophilic Alicyclobacillus acidocaldarius. Acta Crystallogr Sect D-Biological Crystallogr 65: 744-750. doi: 10.1107/S0907444909012773
|
| [54] | Eckert K, Vigouroux A, Lo Leggio L, et al. (2009) Crystal Structures of A. acidocaldarius Endoglucanase Cel9A in Complex with Cello-Oligosaccharides: Strong-1 and-2 Subsites Mimic Cellobiohydrolase Activity. J Mol Biol 394: 61-70. |
| [55] |
Mandelman D, Belaich A, Belaich JP, et al. (2003) X-ray crystal structure of the multidomain endoglucanase Cel9G from Clostridium cellulolyticum complexed with natural and synthetic cello-oligosaccharides. J Bacteriol 185: 4127-4135. doi: 10.1128/JB.185.14.4127-4135.2003
|
| [56] |
Parsiegla G, Belaich A, Belaich JP, et al. (2002) Crystal structure of the cellulase Ce19M enlightens structure/function relationships of the variable catalytic modules in glycoside hydrolases. Biochemistry 41: 11134-11142. doi: 10.1021/bi025816m
|
| [57] |
Sandgren M, Berglund GI, Shaw A, et al. (2004) Crystal complex structures reveal how substrate is bound in the -4 to the +2 binding sites of Humicola grisea cel12A. J Mol Biol 342: 1505-1517. doi: 10.1016/j.jmb.2004.07.098
|
| [58] | Sandgren M, Shaw A, Ropp TH, et al. (2001) The X-ray crystal structure of the Trichoderma reesei family 12 endoglucanase 3, Cel12A, at 1.9 angstrom resolution. J Mol Biol 308: 295-310. |
| [59] | Sandgren M, Gualfetti PJ, Paech C, et al. (2003) The Humicola grisea Cell2A enzyme structure at 1.2 angstrom resolution and the impact of its free cysteine residues on thermal stability. Protein Sci 12: 2782-2793. |
| [60] |
Kitago Y, Karita S, Watanabe N, et al. (2007) Crystal structure of Cel44A, a glycoside hydrolase family 44 endoglucanase from Clostridium thermocellum. J Biol Chem 282: 35703-35711. doi: 10.1074/jbc.M706835200
|
| [61] |
Valjakka J, Rouvinen J (2003) Structure of 20K endoglucanase from Melanocarpus albomyces at 1.8 angstrom resolution. Acta Crystallogr Sect D-Biol Crystallogr 59: 765-768. doi: 10.1107/S0907444903002051
|
| [62] |
Hirvonen M, Papageorgiou AC (2003) Crystal structure of a family 45 endoglucanase from Melanocarpus albomyces: Mechanistic implications based on the free and cellobiose-bound forms. J Mol Biol 329: 403-410. doi: 10.1016/S0022-2836(03)00467-4
|
| [63] |
Parsiegla G, Reverbel-Leroy C, Tardif C, et al. (2000) Crystal structures of the cellulase Ce148F in complex with inhibitors and substrates give insights into its processive action. Biochemistry 39: 11238-11246. doi: 10.1021/bi001139p
|
| [64] |
Sulzenbacher G, Driguez H, Henrissat B, et al. (1996) Structure of the Fusarium oxysporum endoglucanase I with a nonhydrolyzable substrate analogue: Substrate distortion gives rise to the preferred axial orientation for the leaving group. Biochemistry 35: 15280-15287. doi: 10.1021/bi961946h
|
| [65] |
Li JH, Du LK, Wang LS (2010) Glycosidic-Bond Hydrolysis Mechanism Catalyzed by Cellulase Cel7A from Trichoderma reesei: A Comprehensive Theoretical Study by Performing MD, QM, and QM/MM Calculations. J Phys Chem B 114: 15261-15268. doi: 10.1021/jp1064177
|
| [66] |
Mine Y, Fukunaga K, Itoh K, et al. (2003) Enhanced enzyme activity and enantioselectivity of lipases in organic solvents by crown ethers and cyclodextrins. J Biosci Bioeng 95: 441-447. doi: 10.1016/S1389-1723(03)80042-7
|
| [67] |
Payne CM, Bomble Y, Taylor CB, et al. (2011) Multiple Functions of Aromatic-Carbohydrate Interactions in a Processive Cellulase Examined with Molecular Simulation. J Biol Chem 286: 41028-41035. doi: 10.1074/jbc.M111.297713
|
| [68] |
Voutilainen SP, Boer H, Alapuranen M, et al. (2009) Improving the thermostability and activity of Melanocarpus albomyces cellobiohydrolase Cel7B. Appl Microbiol Biotechnol 83: 261-272. doi: 10.1007/s00253-008-1848-9
|
| [69] | Ding H, Xu F (2004) Lignocellulolse Biodegradation, Chapter 9, ACS 154-169. |
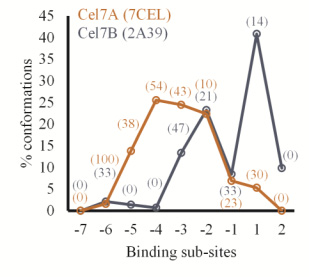
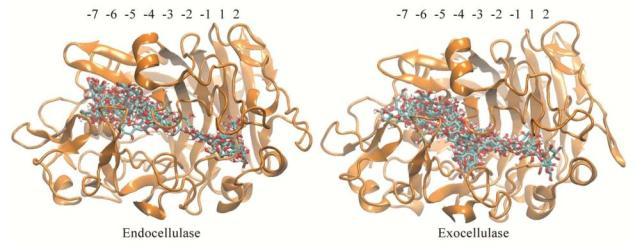
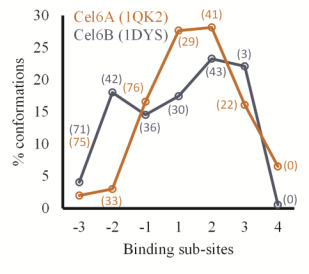
Figures(11) / Tables(1)

Somisetti V. Sambasivarao, David M. Granum, Hua Wang, C. Mark Maupin. Identifying the Enzymatic Mode of Action for Cellulase Enzymes by Means of Docking Calculations and a Machine Learning Algorithm[J]. AIMS Molecular Science, 2014, 1(1): 59-80. doi: 10.3934/molsci.2014.1.59







 DownLoad:
DownLoad: