Lung cancer is a cancer with the fastest growth in the incidence and mortality all over the world, which is an extremely serious threat to human's life and health. Evidences reveal that external environmental factors are the key drivers of lung cancer, such as smoking, radiation exposure and so on. Therefore, it is urgent to explain the mechanism of lung cancer risk due to external environmental factors experimentally and theoretically. However, it is still an open issue regarding how external environment factors affect lung cancer risk. In this paper, we summarize the main mathematical models involved the gene mutations for cancers, and review the application of the models to analyze the mechanism of lung cancer and the risk of lung cancer due to external environmental exposure. In addition, we apply the model described and the epidemiological data to analyze the influence of external environmental factors on lung cancer risk. The result indicates that radiation can cause significantly an increase in the mutation rate of cells, in particular the mutation in stability gene that leads to genomic instability. These studies not only can offer insights into the relationship between external environmental factors and human lung cancer risk, but also can provide theoretical guidance for the prevention and control of lung cancer.
Citation: Lingling Li, Mengyao Shao, Xingshi He, Shanjing Ren, Tianhai Tian. Risk of lung cancer due to external environmental factor and epidemiological data analysis[J]. Mathematical Biosciences and Engineering, 2021, 18(5): 6079-6094. doi: 10.3934/mbe.2021304
Lung cancer is a cancer with the fastest growth in the incidence and mortality all over the world, which is an extremely serious threat to human's life and health. Evidences reveal that external environmental factors are the key drivers of lung cancer, such as smoking, radiation exposure and so on. Therefore, it is urgent to explain the mechanism of lung cancer risk due to external environmental factors experimentally and theoretically. However, it is still an open issue regarding how external environment factors affect lung cancer risk. In this paper, we summarize the main mathematical models involved the gene mutations for cancers, and review the application of the models to analyze the mechanism of lung cancer and the risk of lung cancer due to external environmental exposure. In addition, we apply the model described and the epidemiological data to analyze the influence of external environmental factors on lung cancer risk. The result indicates that radiation can cause significantly an increase in the mutation rate of cells, in particular the mutation in stability gene that leads to genomic instability. These studies not only can offer insights into the relationship between external environmental factors and human lung cancer risk, but also can provide theoretical guidance for the prevention and control of lung cancer.

| [1] |
B. Vogelstein, K. W. Kinzler, Cancer genes and the pathways they control, Nat. Med., 10 (2004), 789–799. doi: 10.1038/nm1087

|
| [2] | D. Hanahan, R. A. Weinberg, The hallmarks of cancer, Cell, 100 (2000), 57–70. |
| [3] |
D. Hanahan, R. A. Weinberg, The hallmarks of cancer: The next generation, Cell, 144 (2011), 646–674. doi: 10.1016/j.cell.2011.02.013
|
| [4] |
A. Amer, A. Nagah, T. T. Tian, X. A. Zhang, Mutation mechanisms of breast cancer among the female population in china, Curr. Bioinform., 15 (2020), 253–259. doi: 10.2174/1574893615666191220141548
|
| [5] |
L. D. Wood, D. W. Parson, S. Jones, J. Lin, T. Sj$\ddot{o}$blom, R. J. Leary, et al., The genomic landscapes of human breast and colorectal cancers, Science, 318 (2007), 1108–1113. doi: 10.1126/science.1145720
|
| [6] | S. Basu, S. Lee, J. Salotti, S. Basu, K. Sakchaisri, X. Xiao, et al., Oncogenic RAS-induced perinuclear signaling complexes requiring KSR1 regulate signal transmission to downstream targets, Cancer Res., 78 (2017), 891–908. |
| [7] |
S. Y. Hong, Y. R. Kao, T. C. Lee, C. W. Wu, Upregulation of e3 ubiquitin ligase cblc enhances egfr dysregulation and signaling in lung adenocarcinoma, Cancer Res., 78 (2018), 4984–4996. doi: 10.1158/0008-5472.CAN-17-3858
|
| [8] |
I. Bozic, T. Antal, H. Ohtsuki, H. Carter, M. A. Nowak, Accumulation of driver and passenger mutations during tumor progression, Proc. Natl. Acad. Sci. U.S.A., 107 (2010), 18545–18550. doi: 10.1073/pnas.1010978107
|
| [9] |
C. Tomasetti, B. Vogelstein, G. Parmigiani, Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor, Proc. Natl. Acad. Sci. U.S.A., 110 (2013), 1999–2004. doi: 10.1073/pnas.1221068110
|
| [10] |
S. Jones, W. D. Chen, G. Parmigiani, F. Diehl, S. D. Markowitz, Comparative lesion sequencing provides insights into tumor evolution, Proc. Natl. Acad. Sci. U.S.A., 105 (2008), 4283–4288. doi: 10.1073/pnas.0712345105
|
| [11] | B. Vogelstein, N. Papadopoulos, V. E. Velculescu, S. Zhou, L. A. Diaz, K. W. Kinzler, Cancer genome landscapes, Science, 339 (2013), 1546–1558. |
| [12] |
P. Armitage, R. Doll, The age distribution of cancer and multi-stage theory of carcinogenesis, Br. J. Cancer, 8 (1954), 1–12. doi: 10.1038/bjc.1954.1
|
| [13] | P. Armitage, R. Doll, A two-stage theory of carcinogenesis in relation to the age distribution of human cancer, Br. J. Cancer, 9 (1957), 161–169. |
| [14] |
A. G. Knudson, Mutation and cancer: statistical study of retinoblastoma, Proc. Natl. Acad. Sci. U.S.A., 68 (1971), 820–823. doi: 10.1073/pnas.68.4.820
|
| [15] |
S. H. Moolgavkar, A. G. Knudson, Mutation and cancer: a model for human carcinogenesis, J. Natl. Cancer. Inst., 66 (1981), 1037–1052. doi: 10.1093/jnci/66.6.1037
|
| [16] | S. H. Moolgavkar, N. E. Day, R. G. Stevens, Two-stage model for carcinogenesis: epidemiology of breast cancer in females, J. Natl. Cancer Inst., 65 (1980), 559–569. |
| [17] |
S. H. Moolgavkar, A. Dewanji, D. J. Venzon, A stochastic two-stage model for cancer risk assessment I: The hazard function and the probability of tumor, Risk Anal., 8 (1988), 383–392. doi: 10.1111/j.1539-6924.1988.tb00502.x
|
| [18] |
R. Meza, J. Jeon, S. H. Moolgavkar, E. G. Luebeck, Age-specific incidence of cancer: phases, transitions, and biological implications, Proc. Natl. Acad. Sci. U.S.A., 105 (2008), 16284–16289. doi: 10.1073/pnas.0801151105
|
| [19] | S. H. Moolgavkar, T. R. Holford, D. T. Levy, C. Y. Kong, M. Foy, L. Clarke, et al., Impact of reduced tobacco smoking on lung cancer mortality in the United States during 1975–2000, J. Natl. Cancer Inst., 104 (2012), 1–7. |
| [20] |
W. R$\ddot{u}$hm, M. Eidemller, J. C. Kaiser, Biologically-based mechanistic models of radiation-related carcinogenesis applied to epidemiological data, Int. J. Radiat. Biol., 93 (2017), 1093–1117. doi: 10.1080/09553002.2017.1310405
|
| [21] |
L. Li, T. Tian, X. Zhang, The impact of radiation on the development of lung cancer, J. Theor. Biol., 428 (2017), 147–152. doi: 10.1016/j.jtbi.2017.06.020
|
| [22] |
B. M. Lang, J. Kuipers, B. Misselwitz, N. Beerenwinkel, Predicting colorectal cancer risk from adenoma detection via a two-type branching process model, PLOS Comput. Biol., 16 (2020), e1007552. doi: 10.1371/journal.pcbi.1007552
|
| [23] |
C. Paterson, H. Clevers, I. Bozic, Mathematical model of colorectal cancer initiation, Proc. Natl. Acad. Sci. U.S.A., 117 (2020), 20681–20688. doi: 10.1073/pnas.2003771117
|
| [24] |
M. A. Nowak, N. L. Komarova, A. Sengupta, P. V. Jallepalli, I. M. Shih, B. Vogelstein, et al., The role of chromosomal instability in tumor initiation, Proc. Natl. Acad. Sci. U.S.A., 99 (2002), 16226–16231. doi: 10.1073/pnas.202617399
|
| [25] |
M. P. Little, E. G. Wright, A stochastic carcinogenesis model incorporating genemic instability fitted to colon cancer data, Math. Biosci., 183 (2003), 111–134. doi: 10.1016/S0025-5564(03)00040-3
|
| [26] | M. P. Little, P. Vineis, G. Li, A stochastic carcinogenesis model incorporating multiple types of genomic instability fitted to colon cancer data, J. Theor. Biol., 254 (2017), 229–238. |
| [27] |
M. P. Little, Cancer models, genomic instability and somatic cellular darwinian evolution, Biol. Direct., 5 (2010), 1–19. doi: 10.1186/1745-6150-5-1
|
| [28] |
S. Z$\ddot{o}$llner, M. E. Sokolnikov, M. Eidem$\ddot{o}$ller, Beyond two-stage models for lung carcinogenesis in the Mayak Workers: implications for plutonium risk, PLoS ONE, 10 (2015), 1–20. doi: 10.1371/journal.pone.0116884
|
| [29] |
M. Eidem$\ddot{u}$ller, E. Holmberg, P. Jacob, M. Lundell, P. Karlsson, Breast cancer risk and possible mechanisms of radiation-induced genomic instability in the Swedish hemangioma cohort after reanalyzed dosimetry, Mutat. Res., 775 (2015), 1–9. doi: 10.1016/j.mrfmmm.2015.03.002
|
| [30] |
L. L. Li, T. H. Tian, X. A. Zhang, L. Pang, Mathematical modeling the pathway of genomic stability in lung cancer, Sci. Rep., 9 (2019), 14136–14144. doi: 10.1038/s41598-019-50500-w
|
| [31] |
W. D. Hazelton, G. Goodman, W. N. Rom, M. Tockman, M. Thornquist, S. H. Moolgavkar, et al., Longitudinal multistage model for lung cancer incidence, mortality, and CT detected indolent and aggressive cancers, Math. Biosci., 240 (2012), 20–34. doi: 10.1016/j.mbs.2012.05.008
|
| [32] |
X. Zhang, R. Simon, Estimating the number of rate limiting genomic changes for human breast cancer, Breast Cancer Res. Treat., 91 (2005), 121–124. doi: 10.1007/s10549-004-5782-y
|
| [33] |
X. Zhang, Y. Fang, Y. Zhao, W. Zheng, Mathematical modeling the pathway of human breast cancer, Math. Biosci., 253 (2014), 25–29. doi: 10.1016/j.mbs.2014.03.011
|
| [34] |
L. Li, T. Tian, X. Zhang, Mutation mechanisms of human breast cancer, J. Comput. Biol., 25 (2018), 1–9. doi: 10.1089/cmb.2018.29010.jh
|
| [35] |
C. Tomasetti, L. Marchionni, M. A. Nowak, G. Parmigiani, B. Vogelstein, Only three driver gene mutations are required for the development of lung and colorectal cancers, Proc. Natl. Acad. Sci. U.S.A., 112 (2015), 118–123. doi: 10.1073/pnas.1421839112
|
| [36] |
N. Castelletti, J. C. Kaiser, C. Simonetto, K. Furukawa, H. Küchenhoff, G.T. Stathopoulos, Risk of lung adenocarcinoma from smoking and radiation arises in distinct molecular pathways, Carcinogenesis, 40 (2019), 1240–1250. doi: 10.1093/carcin/bgz036
|
| [37] |
M. D. Radmacher, R. Simon, Estimation of Tamoxifen's efficiency for preventing the formation and growth of breast tumors, J. Natl. Cancer Inst., 92 (2000), 48–53. doi: 10.1093/jnci/92.1.48
|
| [38] |
L. L. Li, T. T. Tian, X. A. Zhang, Stochastic modelling of multistage carcinogenesis and progression of human lung cancer, J. Theor. Biol., 479 (2019), 81–89. doi: 10.1016/j.jtbi.2019.07.006
|
| [39] |
S. H. Moolgavkar, G. Luebeck, Two-event model for carcinogenesis: biological, mathematical, and statistical considerations, Risk Anal., 10 (1990), 323–341. doi: 10.1111/j.1539-6924.1990.tb01053.x
|
| [40] | E. Parzen, Stochastic processes, in Applied Mathematics, San Francisco: Holden-Day, (1962), 188–299. |
| [41] |
H. J. Clewell, D. W. Quinn, M. E. Andersen, R. B. Conolly, An improved approximation to the exact solution of the two-stage clonal growth model of cancer, Risk Anal., 15 (1995), 467–473. doi: 10.1111/j.1539-6924.1995.tb00339.x
|
| [42] | R. T. Hoogenveen, H. J. Clewell, M. E. Andersen, W. Slob, An alternative exact solution of the two- stage clonal growth model of cancer, Risk Anal., 19 (1999), 9–14. |
| [43] |
K. S. Crump, R. P. Subramaniam, C. B. van Landingham, A numerical solution to the nonhomogeneous two-stage MVK model of cancer, Risk Anal., 25 (2005), 921–926. doi: 10.1111/j.1539-6924.2005.00651.x
|
| [44] | W. Song, S. Powers, Z. Wei, A. H. Yusuf, Substantial contribution of extrinsic risk factors to cancer development, Nature, 529 (2015), 43–47. |
| [45] |
N. L. Komarovaa, A. Senguptac, M. A. Nowak, Mutation cselection networks of cancer initiation: tumor suppressor genes and chromosomal instability, J. Theor. Biol., 223 (2003), 433–450. doi: 10.1016/S0022-5193(03)00120-6
|
| [46] |
M. A. Nowak, F. Michor, Y. Iwasa, Genetic instability and clonal expansion, J. Theor. Biol., 241 (2006), 26–32. doi: 10.1016/j.jtbi.2005.11.012
|
| [47] |
A. D. Asatryan, N. L. Komarova, Evolution of genetic instability in heterogeneous tumors, J. Theor. Biol., 396 (2016), 1–12. doi: 10.1016/j.jtbi.2015.11.028
|
| [48] |
C. M. Choi, K. W. Seo, S. J. Jang, Y. M. Oh, T. S. Shim, W. S. Kim, et al., Chromosomal instability is a risk factor for poor prognosis of adenocarcinoma of the lung: Fluorescence in situ hybridization analysis of paraffin-embedded tissue from Korean patients, Lung Cancer, 64 (2009), 66–70. doi: 10.1016/j.lungcan.2008.07.016
|
| [49] | V. I. Minina, M. Y. Sinitsky, V. G. Druzhinin, A. Fucic, M. L. Bakanova, A. V. Ryzhkova, et al., Chromosome aberrations in peripheral blood lymphocytes of lung cancer patients exposed to radon and air pollution, Eur. J. Cancer Prev., 27 (2016), 6–12. |
| [50] | C. J. Gomes, S. Centuori, J. D. Martinez, Abstract 3509: Overexpression of 14-3-3g contributes to chromosomal instability in human lung cancer, Cancer Res., 74 (2014), 3509–3509. |
| [51] | D. M. Parkin, F. Bray, J. Ferlay, P. Pisani, Global cancer statistics, 2002, CA Cancer J. Clin., 55 (2005), 74–108. |
| [52] |
J. Ferlay, H. R. Shin, F. Bray, D. Forman, C. Mathers, D. M. Parkin, Estimates of worldwide burden of cancer in 2008: globocan 2008, Int. J. Cancer, 127 (2010), 2893–2917. doi: 10.1002/ijc.25516
|
| [53] |
J. C. Martin, N. Lunet, A. G. Marr$\acute{o}$n, C. L. Moyano, N. M. Santander, R. Cleries, et al., Projections in breast and lung cancer mortality among women: a bayesian analysis of 52 countries worldwide, Cancer Res., 78 (2018), 4436–4442. doi: 10.1158/0008-5472.CAN-18-0187
|
| [54] |
L. Walsh, F. Dufey, A. Tschense, M. Schnelzer, B. Grosche, M. Kreuzer, Radon and the risk of cancer mortality-internal Poisson models for the German uranium miners cohort, Health Phys., 99 (2010), 292–300. doi: 10.1097/HP.0b013e3181cd669d
|
| [55] |
I. Zaballa, M. Eidem$\ddot{u}$ller, Mechanistic study on lung cancer mortality after radon exposure in the wismut cohort supports important role of clonal expansion in lung carcinogenesis, Radiat. Environ. Biophys., 55 (2016), 299–315. doi: 10.1007/s00411-016-0659-0
|
| [56] | P. Jacob, R. Meckbach, M. Sokolnikov, V. V. Khokhryakov, E. Vasilenko, Lung cancer risk of Mayak workers: modelling of carcinogenesis and bystander effect, Radiat. Environ. Biophys., 46, (2007), 383–394. |
| [57] |
W. Heidenreich, L. Tomasek, B. Grosche, K. Leuraud, D. Laurier, Lung cancer mortality in the European uranium miners cohort analysed with a biologically based model taking into account radon measurement error, Radiat. Environ. Biophys., 51 (2012), 263–275. doi: 10.1007/s00411-012-0423-z
|
| [58] |
M. Eidem$\ddot{u}$ller, P. Jacob, R. S. D. Lane, S. E. Frost, L. B. Zablotska, Lung cancer mortality (1950–1999) among Eldorado uranium workers: a comparison of models of carcinogenesis and empirical excess risk models, PloS ONE, 7 (2012), e41431. doi: 10.1371/journal.pone.0041431
|
| [59] |
K. Furukawa, D. L. Preston, S. L$\ddot{o}$nn, S. Funamoto, K. Mabuchi, Radiation and smoking effects on lung cancer incidence among atomic bomb survivors, Radiat. Res., 174 (2010), 72–82. doi: 10.1667/RR2083.1
|
| [60] |
J. D. Campbell, A. Alexandrov, J. Kim, J. Wala, A. H. Berger, C. Sekhar, et al., Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas, Nat. Genet., 48 (2016), 607–616. doi: 10.1038/ng.3564
|
| [61] |
K. Wu, X. Zhang, F. Li, D. Xiao, Y. Hou, S. Zhu, et al., Frequent alterations in cytoskeleton remodelling genes in primary and metastatic lung adenocarcinomas, Nat. Commun., 6 (2015), 10131. doi: 10.1038/ncomms10131
|
| [62] | A. F. Brouwer, R. Meza, M. C. Eisenberg, A systematic approach to determining the identifiability of multistage carcinogenesis models, Risk Anal., 37 (2016), 1375–1387. |
| [63] |
T. Alarcn, H. M. Byrne, P. K. Maini, A multiple scale model for tumor growth, Multiscale Model. Sim., 3 (2005), 440–475. doi: 10.1137/040603760
|
| [64] |
M. D. Johnston, C. M. Edwards, W. F. Bodmer, P. K. Maini, S. J. Chapman, Mathematical modeling of cell population dynamics in the colonic crypt and in colorectal cancer, Proc. Natl. Acad. Sci. U.S.A., 104 (2007), 4008–4013. doi: 10.1073/pnas.0611179104
|
| [65] |
A. G. Fletcher, P. J. Murray, P. K. Maini, Multiscale modelling of intestinal crypt organization and carcinogenesis, Math. Models Methods Appl. Sci., 25 (2015), 2563–2585. doi: 10.1142/S0218202515400187
|
| [66] |
R. D. L. Cruz, P. Guerrero, J. Calvo, T. Alarcón, Coarse-graining and hybrid methods for efficient simulation of stochastic multi-scale models of tumour growth, J. Comput. Physics, 350 (2017), 974–991. doi: 10.1016/j.jcp.2017.09.019
|
| [67] | A. R. A. Anderson, P. K. Maini, Mathematical oncology, Bull. Math. Biol., 80 (2018), 945–953. |
| [68] | C. Surulescu, J. Kelkel, On some models for cancer cell migration through tissue networks, Math. Biosci. Eng., 8 (2012), 575–589. |
| [69] |
H. L. Yang, J. Z. Lei, A mathematical model of chromosome recombination-induced drug resistance in cancer therapy, Math. Biosci. Eng., 16 (2019), 7098–7111. doi: 10.3934/mbe.2019356
|
| [70] |
The Cancer Genome Atlas Research Network, Comprehensive genomic characterization of squamous cell lung cancers, Nature, 489 (2012), 519–525. doi: 10.1038/nature11404
|
| [71] |
The Cancer Genome Atlas Research Network, Comprehensive molecular profiling of lung adenocarcinoma, Nature, 511 (2014), 543–550. doi: 10.1038/nature13385
|
| [72] |
G. J. Thomas, The cancer genome atlas research network: A sight to behold, Endocr. Pathol., 25 (2014), 362–365. doi: 10.1007/s12022-014-9345-4
|
| [73] |
N. Misra, E. Szczurek, M. Vingron, Inferring the paths of somatic evolution in cancer, Bioinformatics, 30 (2014), 2456–2463. doi: 10.1093/bioinformatics/btu319
|
| [74] |
D. Ramazzotti, G. Caravagna, L. O. Loohuis, A. Graudenzi, I. Kosunsky, A. Paroni, et al., CAPRI: efficient inference of cancer progression models from cross-sectional data, Bioinformatics, 31 (2015), 3016–3026. doi: 10.1093/bioinformatics/btv296
|
| [75] | G. Caravagna, A. Graudenzi, D. Ramazzotti, L. D. Sano, G. Mauri, V. Moreno, et al., Algorithmic methods to infer the evolutionary trajectories in cancer progression, Proc. Natl. Acad. Sci. U.S.A., 113 (2015), E4025. |
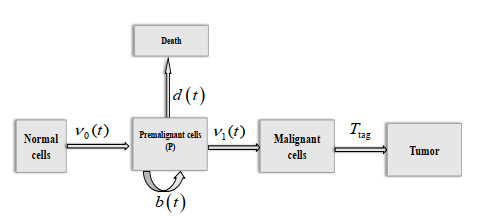
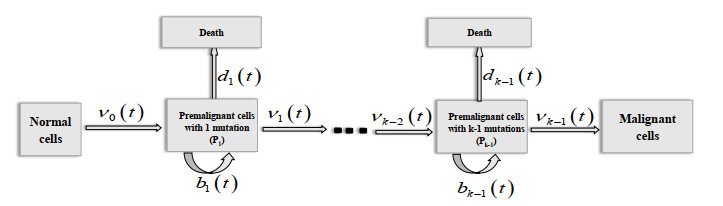
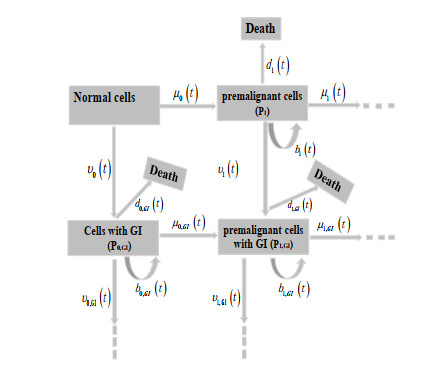
Figures(3) / Tables(2)

Lingling Li, Mengyao Shao, Xingshi He, Shanjing Ren, Tianhai Tian. Risk of lung cancer due to external environmental factor and epidemiological data analysis[J]. Mathematical Biosciences and Engineering, 2021, 18(5): 6079-6094. doi: 10.3934/mbe.2021304







 DownLoad:
DownLoad: