Citation: Enfeng Qi, Can Fu, Ying Zhai, Jianghui Dong. Insights into protease sequence similarities by comparing substrate sequences and phylogenetic dynamics[J]. Mathematical Biosciences and Engineering, 2021, 18(1): 837-850. doi: 10.3934/mbe.2021044

| [1] |
N. D. Rawlings, F. R. Morton, C. Y. Kok, J. Kong, A. J. Barrett, MEROPS: The peptidase database, Nucleic Acids Res., 36 (2008), 320-325. doi: 10.1093/nar/gkn292

|
| [2] |
B. Turk, Targeting proteases: Successes, failures and future prospects, Nat. Rev. Drug Discovery, 5 (2006), 785-799. doi: 10.1038/nrd2092
|
| [3] |
M. Egeblad, Z. Werb, New functions for the matrix metalloproteinases in cancer progression, Nat. Rev. Cancer, 2 (2002), 161-174. doi: 10.1038/nrc745
|
| [4] |
K. Nabeshima, T. Inoue, Y. Shimao, T. Sameshima, Matrix metalloproteinases in tumor invasion: Role for cell migration, Pathol. Int., 52 (2002), 255-264. doi: 10.1046/j.1440-1827.2002.01343.x
|
| [5] |
A. C. Newby, Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates, Cardiovascul. Res., 69 (2006), 614-624. doi: 10.1016/j.cardiores.2005.08.002
|
| [6] |
R. Palmisano, Y. Itoh, Analysis of MMP-dependent cell migration and invasion, Methods Molecul. Biol., 622 (2010), 379-392. doi: 10.1007/978-1-60327-299-5_23
|
| [7] | A. Page-McCaw, A. J. Ewald, Z. Werb, Matrix metalloproteinases and the regulation of tissue remodelling, Nat. Rev. Molecul. Cell Biol., 8 (2007), 221-233. |
| [8] |
O. Julien, J. A. Wells, Caspases and their substrates, Cell Death Diff., 24 (2017), 1380-1389. doi: 10.1038/cdd.2017.44
|
| [9] | X. L. Li, P. Wang, Y. Xie, Protease nexin-1 protects against Alzheimer's disease by regulating the sonic hedgehog signaling pathway, Int. J. Neurosci., (2020), 1-10. |
| [10] | M. A. Slack, S. M. Gordon, Protease activity in vascular disease, Arterioscler. Thromb. Vascul. Biol., 39 (2019), 210-218. |
| [11] |
C. Tomuschat, A. M. O'Donnell, D. Coyle, P. Puri, Increased protease activated receptors in the colon of patients with Hirschsprung's disease, J. Pediatr. Surg., 55 (2020), 1488-1494. doi: 10.1016/j.jpedsurg.2019.11.009
|
| [12] | L. J. Visser, G. N. Medina, H. H. Rabouw, R. J. de Groot, M. A. Langereis, T. de Los Santos, et al., Foot-and-mouth disease virus leader protease cleaves G3BP1 and G3BP2 and inhibits stress granule formation, J. Virol., 93 (2019), 922-918. |
| [13] |
K. Ożegowska, J. Bartkowiak-Wieczorek, A. Bogacz, A. Seremak-Mrozikiewicz, A. J. Duleba, L. Pawelczyk, Relationship between adipocytokines and angiotensin converting enzyme gene insertion/deletion polymorphism in lean women with and without polycystic ovary syndrome, Gynecol. Endocrinology.: Off. J. Int. Soc. Gynecol. Endocrinol., 36 (2020), 496-500. doi: 10.1080/09513590.2019.1695248
|
| [14] |
X. S. Ren, Y. Tong, Y. Qiu, C. Ye, N. Wu, X.Q. Xiong, et al., MiR155-5p in adventitial fibroblasts-derived extracellular vesicles inhibits vascular smooth muscle cell proliferation via suppressing angiotensin-converting enzyme expression, J. Extracell. Vesicles, 9 (2020), 1698795. doi: 10.1080/20013078.2019.1698795
|
| [15] |
I. Schechter, A. Berger, On the size of the active site in proteases. I. Papain. 1967, Biochem. Biophys. Res. Commun., 425 (2012), 497-502. doi: 10.1016/j.bbrc.2012.08.015
|
| [16] |
P. Van Damme, A. Staes, S. Bronsoms, K. Helsens, N. Colaert, E. Timmerman, et al., Complementary positional proteomics for screening substrates of endo and exoproteases, Nat. Methods, 7 (2010), 512-515. doi: 10.1038/nmeth.1469
|
| [17] |
O. Schilling, O. Barré, P. F. Huesgen, C. M. Overall, Proteome-wide analysis of protein carboxy termini: C terminomics, Nat. Methods, 7 (2010), 508-511. doi: 10.1038/nmeth.1467
|
| [18] |
P. Van Damme, S. Maurer-Stroh, K. Plasman, J. Van Durme, N. Colaert, E. Timmerman, et al., Analysis of protein processing by N-terminal proteomics reveals novel species-specific substrate determinants of granzyme B orthologs, Mol. Cell. Proteomics: MCP, 8 (2009), 258-272. doi: 10.1074/mcp.M800060-MCP200
|
| [19] |
S. Mahrus, J. C. Trinidad, D. T. Barkan, A. Sali, A. L. Burlingame, J. A. Wells, Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini, Cell, 134 (2008), 866-876. doi: 10.1016/j.cell.2008.08.012
|
| [20] |
N. D. Rawlings, A. J. Barrett, R. Finn, Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors, Nucleic Acids Res., 44 (2016), 343-350. doi: 10.1093/nar/gkv1118
|
| [21] |
Y. Igarashi, A. Eroshkin, S. Gramatikova, K. Gramatikoff, Y. Zhang, J. W. Smith, et al., CutDB: A proteolytic event database, Nucleic Acids Res., 35 (2007), 546-549. doi: 10.1093/nar/gkl813
|
| [22] |
Y. Igarashi, E. Heureux, K. S. Doctor, P. Talwar, S. Gramatikova, K. Gramatikoff, et al., PMAP: Databases for analyzing proteolytic events and pathways, Nucleic Acids Res., 37 (2009), 611-618. doi: 10.1093/nar/gkn977
|
| [23] |
V. Quesada, G. R. Ordóñez, L. M. Sánchez, X. S. Puente, C. López-Otín, The Degradome database: Mammalian proteases and diseases of proteolysis, Nucleic Acids Res., 37 (2009), 239-243. doi: 10.1093/nar/gkn570
|
| [24] |
A. U. Lüthi, S. J. Martin, The CASBAH: A searchable database of caspase substrates, Cell Death Differ., 14 (2007), 641-650. doi: 10.1038/sj.cdd.4402103
|
| [25] |
K. K. Dey, D. Y. Xie, M. Stephens, A new sequence logo plot to highlight enrichment and depletion, Bmc Bioinf., 19 (2018), 1-9. doi: 10.1186/s12859-017-2006-0
|
| [26] |
G. E. Crooks, G. Hon, J. M. Chandonia, S. E. Brenner, WebLogo: A sequence logo generator, Genome Res., 14 (2004), 1188-1190. doi: 10.1101/gr.849004
|
| [27] |
N. Colaert, K. Helsens, L. Martens, J. L. Vandekerckhove, K. Gevaert, Improved visualization of protein consensus sequences by iceLogo, Nat. Methods, 6 (2009), 786-787. doi: 10.1038/nmeth1109-786
|
| [28] |
M. M. Dix, G.M. Simon, B. F. Cravatt, Global mapping of the topography and magnitude of proteolytic events in apoptosis, Cell, 134 (2008), 679-691. doi: 10.1016/j.cell.2008.06.038
|
| [29] |
J. E. Fuchs, S. von Grafenstein, R. G. Huber, M. A. Margreiter, G. M. Spitzer, H. G. Wallnoefer, et al., Cleavage entropy as quantitative measure of protease specificity, PLoS Comput. Biol., 9 (2013), 1003007. doi: 10.1371/journal.pcbi.1003007
|
| [30] |
J. E. Fuchs, S. von Grafenstein, R. G. Huber, C. Kramer, K. R. Liedl, Substrate-driven mapping of the degradome by comparison of sequence logos, PLoS Comput. Biol., 9 (2013), 1003353. doi: 10.1371/journal.pcbi.1003353
|
| [31] |
E. Qi, D. Wang, Y. Li, G. Li, Z. Su, Revealing favorable and unfavorable residues in cooperative positions in protease cleavage sites, Biochem. Biophys. Res. Commun., 519 (2019), 714-720. doi: 10.1016/j.bbrc.2019.09.056
|
| [32] |
E. F. Qi, D. Y. Wang, B. Gao, Y. Li, G. J. Li, Block-based characterization of protease specificity from substrate sequence profile, Bmc Bioinf., 18 (2017), 438. doi: 10.1186/s12859-017-1851-1
|
| [33] |
J. Song, H. Tan, A. J. Perry, T. Akutsu, G. I. Webb, J. C. Whisstock, et al., PROSPER: An integrated feature-based tool for predicting protease substrate cleavage sites, PloS one, 7 (2012), 50300. doi: 10.1371/journal.pone.0050300
|
| [34] |
J. Verspurten, K. Gevaert, W. Declercq, P. Vandenabeele, SitePredicting the cleavage of proteinase substrates, Trends Biochem. Sci., 34 (2009), 319-323. doi: 10.1016/j.tibs.2009.04.001
|
| [35] |
Z. Zhang, S. Schwartz, L. Wagner, W. Miller, A greedy algorithm for aligning DNA sequences, J. Comput. Biol.: J. Comput. Mol. Cell Biol., 7 (2000), 203-214. doi: 10.1089/10665270050081478
|
| [36] |
C. Spearman, The proof and measurement of association between two things, Am. J. Psychol., 100 (1987), 441-471. doi: 10.2307/1422689
|
| [37] |
I. Letunic, P. Bork, Interactive Tree Of Life v2: Online annotation and display of phylogenetic trees made easy, Nucleic Acids Res., 39 (2011), 475-478. doi: 10.1093/nar/gkq818
|
| [38] |
N. M. Ng, R. N. Pike, S. E. Boyd, Subsite cooperativity in protease specificity, Biol. Chem., 390 (2009), 401-407. doi: 10.1515/BC.2009.065
|
| [39] |
H. R. Stennicke, M. RENATUS, M. MELDAL, G. S. SALVESEN, Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8, Biochem. J., 350 (2000), 563-568. doi: 10.1042/bj3500563
|
| [40] |
Y. Choe, F. Leonetti, D. C. Greenbaum, F. Lecaille, M. Bogyo, D. Brömme, et al., Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities, J. Biol. Chem., 281 (2006), 12824-12832. doi: 10.1074/jbc.M513331200
|
| [41] |
S. Elamouri, H. Zhu, J. Yu, R. A. Marr, I. M. Verma, M. S. Kindy, Neprilysin: An enzyme candidate to slow the progression of Alzheimer's disease, Am. J. Pathol., 172 (2008), 1342-1354. doi: 10.2353/ajpath.2008.070620
|
| [42] |
M. Eguiluz, F. Kulcheski, R. Margis, F. Guzman, De novo assembly of vriesea carinata leaf transcriptome to identify candidate cysteine-proteases, Gene, 691 (2019), 96-105. doi: 10.1016/j.gene.2018.12.053
|
 mbe-18-01-044-Table S1-supplementary.pdf mbe-18-01-044-Table S1-supplementary.pdf |
 |
| mbe-18-01-044-Additional file 1- supplementary.xlsx |
|
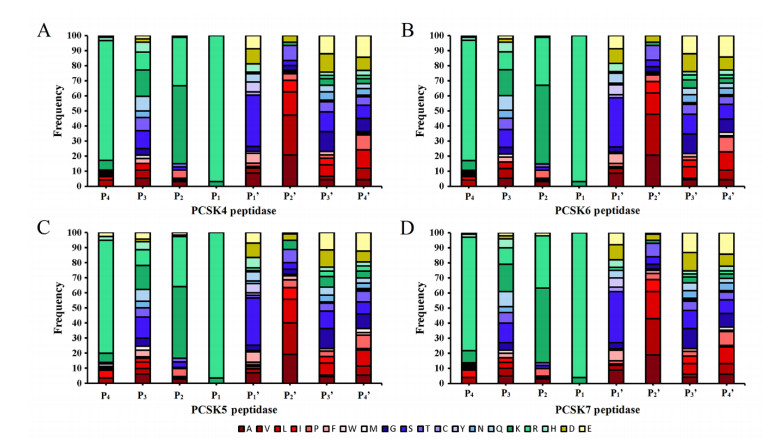
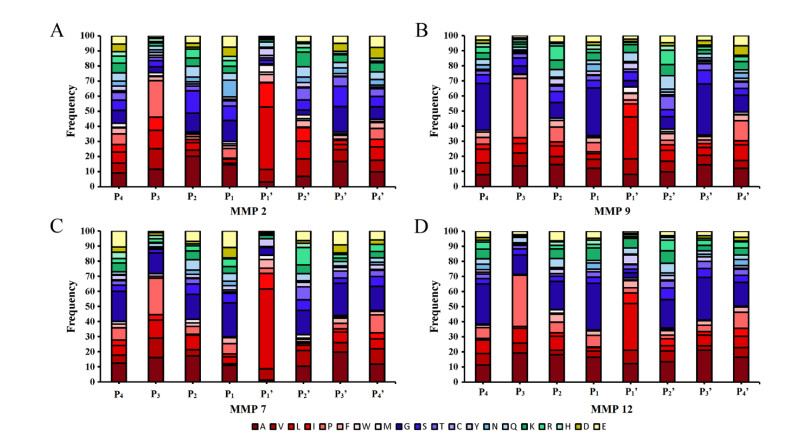
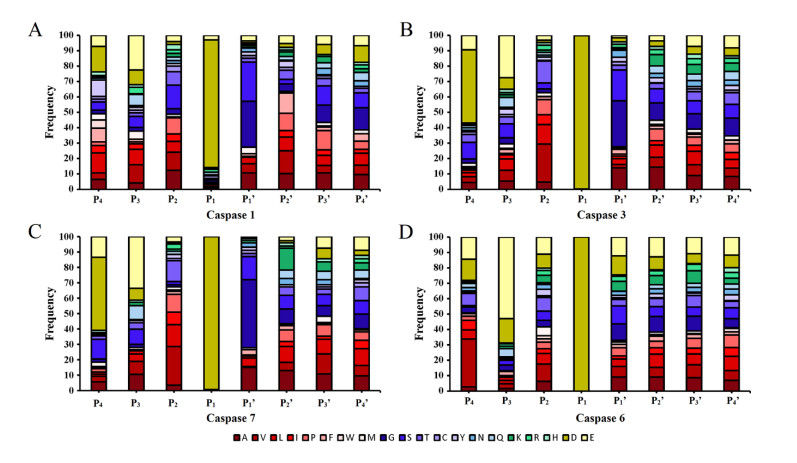
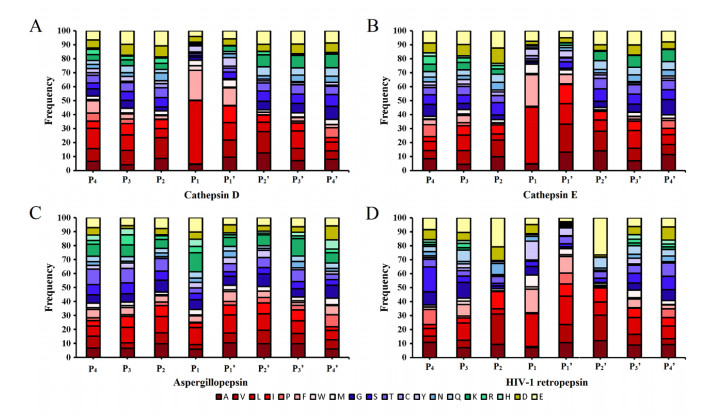
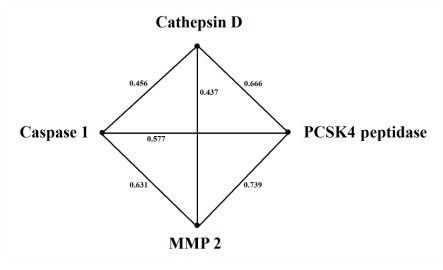
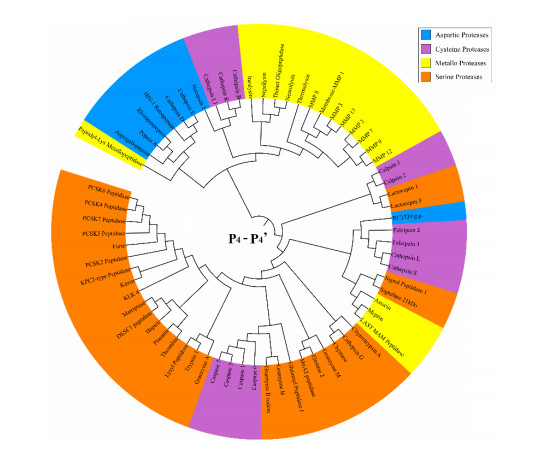
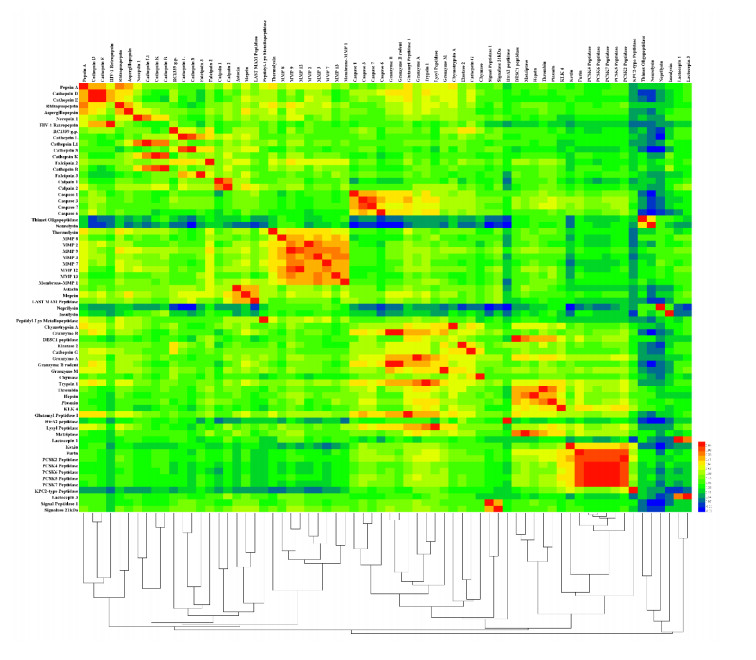
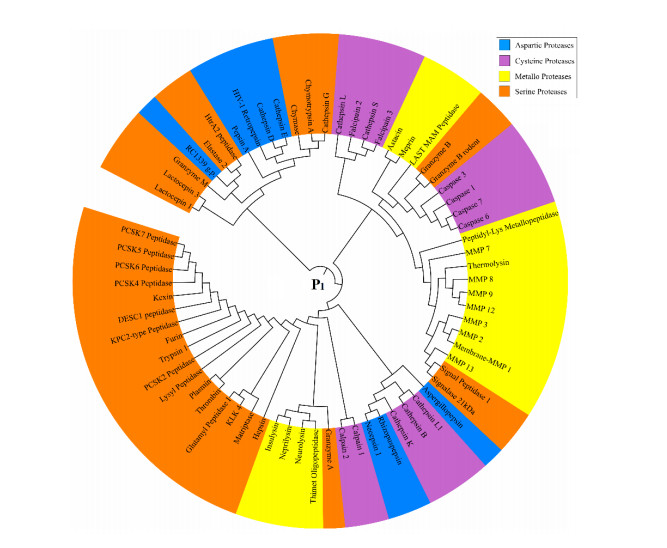
Figures(8)

Enfeng Qi, Can Fu, Ying Zhai, Jianghui Dong. Insights into protease sequence similarities by comparing substrate sequences and phylogenetic dynamics[J]. Mathematical Biosciences and Engineering, 2021, 18(1): 837-850. doi: 10.3934/mbe.2021044







 DownLoad:
DownLoad: