Citation: William A. Toscano, Jr., Hitakshi Sehgal, Emily Yang, Lindsey Spaude, A. Frank Bettmann. Environment-Gene interaction in common complex diseases: New approaches[J]. AIMS Molecular Science, 2014, 1(4): 126-140. doi: 10.3934/molsci.2014.4.126

| [1] |
Nakamura J, Mutlu E, Sharma V, et al. (2014) The Endogenous Exposome. DNA Repair (Amst) 19: 3-13. doi: 10.1016/j.dnarep.2014.03.031

|
| [2] |
Wild CP (2012) The exposome: From concept to utility. Int J Epidemiol 41: 24-32. doi: 10.1093/ije/dyr236
|
| [3] | EPA, Persistent Organic Pollutants: A Global Issue, A Global Response. EPA, 2009. Available from: http://www2.epa.gov/international-cooperation/persistent-organic-pollutants-global-issue-global-response#pops (Accessed July 17, 2014). |
| [4] |
Magliano DJ, Loh VH, Harding JL, et al. (2014) Persistent Organic Pollutants and Diabetes: A Review of the Epidemiological Evidence. Diabetes Meab 40: 1-14. doi: 10.1016/j.diabet.2013.09.006
|
| [5] |
Myre M, Imbeault P (2014) Persistent Organic Pllutants Meet Adipose Tissue Hypoxia: Does Cross-talk Contribute to Obesity. Obes Res 15: 19-28. doi: 10.1111/obr.12086
|
| [6] |
Taylor KW, Novak RF, Anderson HA, et al. (2013) Evaluation of the Association Between Persistent Organic pollutants (POPs) and diabetes in Epidemiological Studies: A National Toxicology Program Workshop Review. Environ Health Perspect 121: 774-783. doi: 10.1289/ehp.1205502
|
| [7] |
Gascon M, Morales E, Sunyer J, et al. (2013) Effects of Persistent Organic Pollutants on the Developing Respiratory and Immune Systems: A Systematic Review. Environ Int 52: 51-65. doi: 10.1016/j.envint.2012.11.005
|
| [8] |
Li QQ, Loganath A, Chong YS, et al. (2006) Persistent Organic Pollutants and Adverse Health Effects in Humans. J Toxicol Env Health Part A 69: 1987-2005. doi: 10.1080/15287390600751447
|
| [9] |
Zielinski I, Tsui I-C (1995) Cystic Fibrosis: Genotypic and Phenotypic Variations. Ann, Rev Genet 29: 777-807. doi: 10.1146/annurev.ge.29.120195.004021
|
| [10] | Braun A, Roth R, McGinnis MJ (2003) Technology challenges in screening single gene disorders. Eur J Pediatr 162: S 13-16. |
| [11] | WHO (2014) World Health Statistics 2014. Geneva, Switzerland: World Health Organization, 180. |
| [12] | Russell JY (2010) A Philosophical Framework for an Open and Critical Transdisciplinary Inquuiry. In: Brown VA, Harris JA, Russell JY, editors. Tackling Wicked Problems Through the Transdisciplinary Imagination. NY, NY: Earthscan, 31-60. |
| [13] | Stokols D, Hall KL, Vogel AL (2013) Transdisciplinary Public Health: Definitions, Core Characteristics, and Strategies for Success. In: Haire-Joshu D, McBride TD, editors. Transdisciplinary Public Health: Research, Education, and Practice. San Francisco, CA: Jossey-Bass, 1-30. |
| [14] |
Jacobs JA, Frickel S (2009) Interdisciplinarity: A Critical Assessment. Annu Rev Sociol 35: 43-65. doi: 10.1146/annurev-soc-070308-115954
|
| [15] | Klein JT (2008) Evaluation of Interdisciplinary and Transdisciplinary Research - A Literature Review. Amer J Prev Med Suppl 2 35: S-116-S-123. |
| [16] | Hays TC (2008) The Science of Team Science - Commentary on Measurements of Scientific Readiness. Amer J Prev Med Suppl 2 35: S-193-S195. |
| [17] | DE (1997) Pasteur's Quadrant: Basic Science and Technological Innovation: Brookings Institution, Washington, DC. |
| [18] | NRC (2014) Convergence: Facilitating Transdisciplinary Integration of Life Sciences, Physical Sciences, Engineering and Beyond. Washington, DC: National Academies Press, 137. |
| [19] | Roche M, Glick TF (1944) Thomas Edison and Basic Science, Edison: Myths and Reality. Arbor: 39-50. |
| [20] | Duggan MB (2014) Prevention of childhood malnutrition: immensity of the challenge and variety of strategies. Paediatr Int Child Health 32: 190-203. |
| [21] |
Blackburn GL (2001) Pasteur's Quadrant and Malnutrition. Nature 409: 397-401. doi: 10.1038/35053187
|
| [22] |
Verbeke W (2005) Consumer acceptance of functional foods: Socio-demographic, cognitive and attitudinal determinants. Food Quality and Preference 16: 45-57. doi: 10.1016/j.foodqual.2004.01.001
|
| [23] | Bhutta ZA, Darmstadt GL (2014) A Role for Science Investments in Advancing Newborn Health. Science Translational Medicine 6: 253-258. |
| [24] | Childress DS (1999) Working in Pasteur's Quadrant. J Rehabilitation Res Dev 36: xi-xii. |
| [25] | De Luigi AJ, Cooper RA (2014) Adaptive sports technology and biomechanics: prosthetics. PM R 8 Suppl.: S40-S57. |
| [26] |
Hardbower DM, Peek RM, Jr., Wilson KT (2014) At the Bench: Helicobacter pylori, dysregulated host responses, DNA damage, and gastric cancer. J Leukoc Biol 96: 201-212. doi: 10.1189/jlb.4BT0214-099R
|
| [27] |
Dalal RS, Moss SF (2014) At the Bedside: Helicobacter pylori, dysregulated host responses, DNA damage, and gastric cancer. J Leukoc Biol 96: 213-224. doi: 10.1189/jlb.4BT0214-100R
|
| [28] | Nobel Committee, The Nobel Prize in Physiology or Medicine Prize Announcement. The Nobel Committee for Physiology or Medicine,2005. Available from:http://www.nobelprize.org/nobel_prizes/medicine/laureates/2005/announcement.html. |
| [29] | Konturek SJ, Konturek PC, Brzozowski T, et al. (2005) From nerves and hormones to bacteria in the stomach; Nobel prize for achievements in gastrology during last century. J Physiol Pharmacol 56: 507-536. |
| [30] |
Marshall B (2006) Helicobacter connections. Chem Med Chem 1: 783-802. doi: 10.1002/cmdc.200600153
|
| [31] | Woo KT, Lau YK, Yap HK, et al. (2006) 3rd College of Physicians' lecture--translational research: From bench to bedside and from bedside to bench; incorporating a clinical research journey in IgA nephritis (1976 to 2006). Ann Acad Med Singapore 35: 735-741. |
| [32] |
Hamilton JG, Edwards HM, Khoury MJ, et al. (2014) Cancer screening and genetics: a tale of two paradigms. Cancer Epidemiol Biomarkers Prev 23: 909-916. doi: 10.1158/1055-9965.EPI-13-1016
|
| [33] | Preston R (2000) The Genome Warrier. The New Yorker 76: 66-83. |
| [34] | Tchounwou PB, Toscano WA (2011) Environmental Epidemiology and Human Health-Biomarkers of Disease and Genetic Susceptibility In: Nriagu JO, editor. Encyclopedia of Environmentl Health Sciences. San Diego, CA: Elsevier, 357-366. |
| [35] |
Teo S, Morgan M, Stirling D, et al. (2000) Assessment of the In Vitro and In Vivo Genotoxicity of Thalomid (Thalidomide). Teratog Carcinog Mutagen 20: 301-311. doi: 10.1002/1520-6866(2000)20:5<301::AID-TCM6>3.0.CO;2-2
|
| [36] |
Portin P (2002) Historical Development of the Concept of the Gene. J Med Philosophy 27: 257-286. doi: 10.1076/jmep.27.3.257.2980
|
| [37] | Crick FHC (1958) Central Dogma of Molecular Biology. Symp Soc Exp Biol XII 12: 139-173. |
| [38] |
Crick FHC (1970) Central Dogma of Molecular Biology. Nature (London) 227: 561-563. doi: 10.1038/227561a0
|
| [39] |
Hahn WC, Weinberg RA (2002) Modeling the Molecular Circuitry of Cancer. Nat Rec Cancer 2: 331-341. doi: 10.1038/nrc795
|
| [40] | Hocquette JF (2005) Where are we in genomics? J Physiol Pharmacol Suppl 3: 37-70. |
| [41] |
Sharma S, Kelly TK, Jones PA (2010) Epigenetics in cancer. Carcinogenesis 31: 27-36. doi: 10.1093/carcin/bgp220
|
| [42] |
Long CR, Westhusin ME, Golding MC (2014) Reshaping the transcriptional frontier: epigenetics and somatic cell nuclear transfer. Mol Reprod Dev 81: 183-193. doi: 10.1002/mrd.22271
|
| [43] | Nilsson E, Skinner MK (2014) Definitions and History of Generational Epigeneti Inheritance. In: Tollefsbol TO, editor. Translational Epigenetics: Evidence and Debate. San Diego, CA: Academic Press, 11-24. |
| [44] | Hoile SP, Lillycrop KA, Grenfel LR, et al. (2014) Phenotypic and Epigenetic Inheritance Across Multiple Generations in Mammals through the Female Line. In: Tollefsbol TO, editor. Transgenerational Epigenetics: Evidence and Debate. San Diego, CA: Academic Press, 269-278. |
| [45] | Mashoodh R, Champagne FA (2014) Paternal Epigenetic Inheritance. In: Tollefsbol TO, editor. Transgenerational Epigenetics: Evidence and Debate, 231-236. |
| [46] | Toscano WA, Oehlke KP (2011) Nutrigenomics: A New Frontier in Environmental Health Sciences. In: Nriagu JO, editor. Encyclopedia of Environmental Health Burlington, VT: Elsevier, 202-205. |
| [47] | Toscano WA, Oehlke KP, Kafouri R (2010) An Environmental Systems Biology Approach to the Study of Asthma. In: Pawnkar R, Holgate S, Rosenwasser LJ, editors. Allergy Frontiers: Future Perspectives. Springer: NY, NY, 239-252. |
| [48] | Toscano WAJ, Lee P, Oehlke KP (2008) The Role of Environmental Health Research in Understanding Chronic Conditions. Minn Physician 21: 27-28. |
| [49] |
McLachlan JA (2001) Environmental Signaling: What Embryos and Evolution Teach us about Endocrine Disrupting Chemicals. Endocr Rev 22: 319-341. doi: 10.1210/edrv.22.3.0432
|
| [50] |
Vandenberg LN (2014) Non-monotonic Dose Responses in Studies of Endocrine Disrupting Chemicals: Bisphenol-A as a Case Study. Dose-Response 12: 259-276. doi: 10.2203/dose-response.13-020.Vandenberg
|
| [51] |
Vandenberg LN, Colborn T, Hayes TB, et al. (2012) Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev 33: 378-455. doi: 10.1210/er.2011-1050
|
| [52] | Vandenberg LN, Erlich S, Belcher SM, et al. (2013) Low-dose Effects of Bisphenol-A: An Integrated review of in vitro, Laboratory, Animal, and Epidemiology Studies. Endocrine Disruptors 1: 1-20. |
| [53] | CDC (2014) National Diabetes Statistics Report, 2014. Center for Disease Control, Atlanta, GA. |
| [54] |
Misra A, Bhardwai S (2014) Obesity and the Metbolic Syndrome in Developing Countries: Focus on South Asians. Nestle Nutr Inst Workshop Ser 78: 133-140. doi: 10.1159/000354952
|
| [55] |
Chase K, Sharma RP (2013) Epigenetic Developmental Programs and Adipogenesis - Implications for Psychotropic Induced Obesity. Epigenetics 8: 1133-1140. doi: 10.4161/epi.26027
|
| [56] | Janesick A, Blumberg B (2011) Minireview: PPARγ as the target of obesogens. J Ster Biochem & Mol Biol 127: 4-8. |
| [57] | Janesick A, Blumberg B (2011) The role of environmental obesogens in the obesity epidemic. Obesity before birth: Springer. 383-399. |
| [58] | Kishida K, Shimomura H, Nishizawa N, et al. (2001) Enhancement of the aquaporin adipose gene expression by a peroxisome proliferator-activated receptor gamma. J Biol Chem 276: 48572-48579. |
| [59] |
Grun F, Blumberg B (2009) Minireview: the case for obesogens. Mol Endocrinol 23: 1127-1134. doi: 10.1210/me.2008-0485
|
| [60] |
Kemper MF, Stirone C, N. KD, et al. (2014) Genomic and non-genomic regulation of PGC1 isoforms by estrogen to increase cerebral vascular mitochondrial biogenesis and reactive oxygen species. Eur J Pharmacol 723: 322-329. doi: 10.1016/j.ejphar.2013.11.009
|
| [61] |
Riu A, Grimaldi M, le Maire A, et al. (2011) Peroxisome Proliferator-Activated Receptor γ Is a Target for Halogenated Analogs of Bisphenol A. Environ Health Perspect 119: 1227-1232. doi: 10.1289/ehp.1003328
|
| [62] |
Swedenborg E, Ruegg J, Mäkelä S, et al. (2009) Endocrine disruptive chemicals: mechanisms of action and involvement in metabolic disorders. J Mol Endovrinol 43: 1-10. doi: 10.1677/JME-08-0132
|
| [63] |
Willy PJ, Murray IR, Busch BB, et al. (2004) Regulation of PPARγ coactivator 1α (PGC-1α) signaling by an estrogen-related receptor α (ERRα) ligand. Proc Natl Acad Sci (USA) 101: 8912-8917. doi: 10.1073/pnas.0401420101
|
| [64] | Alaynick WA (2006) Nuclear Receptors, Mitochondria and Lipid Metabolism. Mitochondrion 8: 329-337. |
| [65] |
Koo S-H, Satoh H, Herzig S, et al. (2004) PGC-1 Promotes Insulin Resistance in Liver through PPAR-alpha-dependent Induction of TRB-3. Nature Med 10: 530-534. doi: 10.1038/nm1044
|
| [66] |
Sargis RM, Johnson DN, Choudhury RA, et al. (2010) Environmental endocrine disruptors promote adipogenesis in the 3T3-L1 cell line through glucocorticoid receptor activation. Obesity 18: 1283-1288. doi: 10.1038/oby.2009.419
|
| [67] |
Unterberger C, Stapls KJ, Smallie T, et al. (2008) Role of STAT3 in Glucocorticoid-induced Expression of the Human IL-10 Gene. Mol Immunol 45: 3230-3237. doi: 10.1016/j.molimm.2008.02.020
|
| [68] |
Tanaka T, Nobuiaki Y, Kishimoto T, et al. (1997) Defective Adipocyte Differentiation in Mice Lacking the C/EBP or C/EBP Gene. EMBO J 16: 7432-7443. doi: 10.1093/emboj/16.24.7432
|
| [69] |
Berger J, Tanen M, Elbrecht A, et al. (2001) Peroxisome Proliferator-activated Receptor-γ Ligands Inhibit Adipocyte 11β-Hydroxysteroid Dehydrogenase Type 1 Expression and Activity. J Biol Chem 276: 12629-12635. doi: 10.1074/jbc.M003592200
|
| [70] |
Fukazawa H, Hoshino K, Shiozawa T, et al. (2001) Identification and Quantification of Chlorinated Bisphenol A in Wastewater from Wastepaper Recycling Plants. Chemosphere 44: 973-979. doi: 10.1016/S0045-6535(00)00507-5
|
| [71] |
Guerra P, Eljarrat E, Barcelo D (2010) Simultaneous Determination Hexabromocyclododecane, Tetrabromobisphenol A, and Related Compounds in Sewage Sludge and Sediment tsamples from the Ebro River Basin (Spain). Anal Bianal Chem 397: 2817-2824. doi: 10.1007/s00216-010-3670-3
|
| [72] | Kolpin DW, Furlong ET, Meyer MT, et al. (2002) Pharmaceuticals, Hormones, and Other Organic Wastewater Contaminants in U.S. Streams, 1999-2000: A National Reconnaissance. Environ Sci Technol 36: 1202-1211. |
| [73] |
Korshin G, Kim J, Gan L (2006) Comparative Study of Reactions of Endocrine Eisruptors, Bisphenol-A and Diethylstilbestrol in Electrochemical Treatment and Chlorination. Water Res 40: 1070-1078. doi: 10.1016/j.watres.2006.01.003
|
| [74] |
Gallard H, Leclercq A, Croué J-P (2004) Chlorination of Bisphenol A: Kinetics and By-products Formation. Chemosphere 56: 465-473. doi: 10.1016/j.chemosphere.2004.03.001
|
| [75] | Levian C, Ruiz E, Yang X (2014) The Pathogenesis of Obesity from a Genomic and Systems Biology Perspective. Yale J Biol Med 87: 113-126. |
| [76] |
Zhao S, Iyengar R (2012) Systems pharmacology: Network analysis to identify multiscale mechanisms of drug action. Ann Rev Pharmacol Toxicol 52: 505-521. doi: 10.1146/annurev-pharmtox-010611-134520
|
| [77] |
Toscano WA, Oehlke KP (2005) Systems Biology: New Approaches to Old Environmental Health Problems. Int J Environ Res Public Health 2: 4-9. doi: 10.3390/ijerph2005010004
|
| [78] | Leischow SJ, Best A, Trochim WM, et al. (2008) Systems Thinking to Improve the Public's Health. Amer J Prev Med Suppl 2 35: S-196 - S-203. |
| [79] |
Cox J, Mann M (2011) Quantitative, High-Resolution Proteomics for Data-Driven Systems Biology. Annu Rev Biochem 80: 273-299. doi: 10.1146/annurev-biochem-061308-093216
|
| [80] |
Li H, Deng H (2010) Systems Genetics, Bioinformatics and eQTL Mapping. Genetica 138: 915-924. doi: 10.1007/s10709-010-9480-x
|
| [81] |
Majithia AR, Flannick J, Shahanian P, et al. (2014) Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc Natl Acad Sci (USA) 111: 13127-13132. doi: 10.1073/pnas.1410428111
|
| [82] |
Hoffman S (2011) Computational Analysis of High Throughput Sequencing Data. Methods Mol Biol 719: 199-217. doi: 10.1007/978-1-61779-027-0_9
|
| [83] | Dondorp WJ, de Wert GM (2013) The Thousand-Dollar Genome: An Ethical Exploration. Eur J Hum Genet Suppl 1: S6-S-26. |
| [84] | Zhang L, Kim S (2014) Learning Gene Networks under SNP Perturbations using eQTL Datasets. PLoS Comput Biol 10: e1003420. |
| [85] |
Meaburn E, Schulz R (2012) Next generation sequencing in epigenetics: insights and challenges. Semin Cell Dev Biol 23: 192-199. doi: 10.1016/j.semcdb.2011.10.010
|
| [86] |
Pellegrini M, Ferrri R (2012) Epigenetic analysis: ChIP-chip and ChIP-seq. Methods Mol Biol 802: 377-387. doi: 10.1007/978-1-61779-400-1_25
|
| [87] |
Ku CS, Naidoo N, Wu M, et al. (2011) Studying the epigenome using next generation sequencing. J Med Genet 48: 721-730. doi: 10.1136/jmedgenet-2011-100242
|
| [88] |
Patel CJ, Battacharya J, Butte AJ (2010) An Environment-Wide Association Study (EWAS) on Type 2 Diabetes Mellitus. PLoS One 5: e10746. doi: 10.1371/journal.pone.0010746
|
| [89] |
Manolio TA, Brooks LD, Collins F (2008) A HapMap harvest of insights into the genetics of common disease. J Clin Invest 118: 1590-1605. doi: 10.1172/JCI34772
|
| [90] | Zeggini E, Weeden MN, Lindgren CM, et al. Replication of Genome-Wide Association Signals in UK Samples Reveals Risk Loci for Type 2 Diabetes. Science (Washington) 316: 1336-1341. |
| [91] | Ioannidis J, Loy EY, Poulton R, et al. (2009) Researching Nongenetic Determinants of Disease A Comparison and Proposed Unification. Sci Transl Med 1: 7ps8. |
| [92] |
Davis AP, Rosenstein MC, Wiegers TC, et al. (2011) DiseaseComps: a metric that discovers similar diseases based upon common toxicogenomic profiles at CTD. Bioinformation 7: 154-156. doi: 10.6026/97320630007154
|
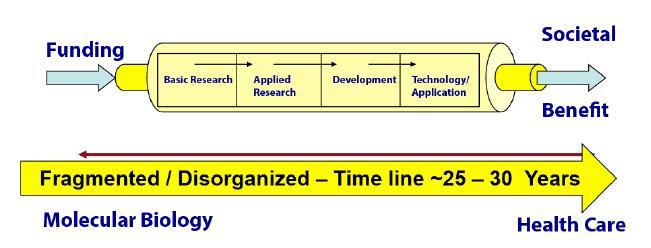

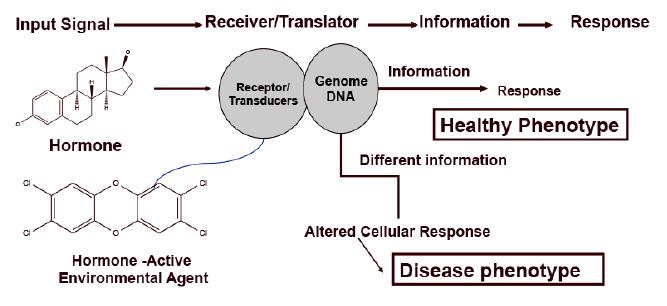
Figures(3)

William A. Toscano, Jr., Hitakshi Sehgal, Emily Yang, Lindsey Spaude, A. Frank Bettmann. Environment-Gene interaction in common complex diseases: New approaches[J]. AIMS Molecular Science, 2014, 1(4): 126-140. doi: 10.3934/molsci.2014.4.126







 DownLoad:
DownLoad: