Citation: Joanna L. Richens, Jonathan P. Bramble, Hannah L. Spencer, Fiona Cantlay, Molly Butler, Paul O’Shea. Towards defining the Mechanisms of Alzheimer’s disease based on a contextual analysis of molecular pathways[J]. AIMS Genetics, 2016, 3(1): 25-48. doi: 10.3934/genet.2016.1.25

| [1] |
Strimbu K, Tavel JA (2010) What are Biomarkers? Curr Opin HIV AIDS 5: 463-466. doi: 10.1097/COH.0b013e32833ed177

|
| [2] |
Robinson DH, Toledo AH (2012) Historical development of modern anesthesia. J Invest Surg 25: 141-149. doi: 10.3109/08941939.2012.690328
|
| [3] | Pitt D, Aubin JM (2012) Joseph Lister: father of modern surgery. Can J Surg 55: E8-9. |
| [4] | Hsu JL (2013) A brief history of vaccines: smallpox to the present. S D Med Spec no: 33-37. |
| [5] |
Blevins SM, Bronze MS (2010) Robert Koch and the 'golden age' of bacteriology. Int J Infect Dis 14: e744-751. doi: 10.1016/j.ijid.2009.12.003
|
| [6] |
Biomarkers Definitions Working Group (2001) Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 69: 89-95. doi: 10.1067/mcp.2001.113989
|
| [7] |
O'Donnell MJ, Xavier D, Liu L, et al. (2010) Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. Lancet 376: 112-123. doi: 10.1016/S0140-6736(10)60834-3
|
| [8] |
Richens JL, Urbanowicz RA, Lunt EA, et al. (2009) Systems biology coupled with label-free high-throughput detection as a novel approach for diagnosis of chronic obstructive pulmonary disease. Respir Res 10: 29. doi: 10.1186/1465-9921-10-29
|
| [9] |
Song IU, Chung YA, Chung SW, et al. (2014) Early diagnosis of Alzheimer's disease and Parkinson's disease associated with dementia using cerebral perfusion SPECT. Dement Geriatr Cogn Disord 37: 276-285. doi: 10.1159/000357128
|
| [10] |
Perazella MA (2015) The Urine Sediment as a Biomarker of Kidney Disease. Am J Kidney Dis 66: 748-755. doi: 10.1053/j.ajkd.2015.02.342
|
| [11] | Felton HT, Derrick JB, Swartz DP (1964) A Simple Immunologic Test For Pregnancy. Can Med Assoc J 91: 996-1000. |
| [12] |
Slamon DJ, Clark GM, Wong SG, et al. (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235: 177-182. doi: 10.1126/science.3798106
|
| [13] | Puglisi F, Fontanella C, Amoroso V, et al. (2015) Current challenges in HER2-positive breast cancer. Crit Rev Oncol Hematol 98: 211-221. |
| [14] |
Anderson NL, Ptolemy AS, Rifai N (2013) The riddle of protein diagnostics: future bleak or bright? Clin Chem 59: 194-197. doi: 10.1373/clinchem.2012.184705
|
| [15] |
Mayeux R (2004) Biomarkers: potential uses and limitations. NeuroRx 1: 182-188. doi: 10.1602/neurorx.1.2.182
|
| [16] |
Anderson NL, Anderson NG (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics 1: 845-867. doi: 10.1074/mcp.R200007-MCP200
|
| [17] |
Krebs HA (1950) Chemical composition of blood plasma and serum. Annu Rev Biochem 19: 409-430. doi: 10.1146/annurev.bi.19.070150.002205
|
| [18] |
Richens JL, Lunt EA, Sanger D, et al. (2009) Avoiding nonspecific interactions in studies of the plasma proteome: practical solutions to prevention of nonspecific interactions for label-free detection of low-abundance plasma proteins. J Proteome Res 8: 5103-5110. doi: 10.1021/pr900487y
|
| [19] |
Drucker E, Krapfenbauer K (2013) Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA J 4: 7. doi: 10.1186/1878-5085-4-7
|
| [20] |
Lim LS, Sherin K (2008) Screening for prostate cancer in U.S. men ACPM position statement on preventive practice. Am J Prev Med 34: 164-170. doi: 10.1016/j.amepre.2007.10.003
|
| [21] |
Prince M, Bryce R, Albanese E, et al. (2013) The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement 9: 63-75 e62. doi: 10.1016/j.jalz.2012.11.007
|
| [22] |
Galvin JE, Sadowsky CH (2012) Practical guidelines for the recognition and diagnosis of dementia. J Am Board Fam Med 25: 367-382. doi: 10.3122/jabfm.2012.03.100181
|
| [23] |
Bekris LM, Yu CE, Bird TD, et al. (2010) Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 23: 213-227. doi: 10.1177/0891988710383571
|
| [24] |
Wu L, Rosa-Neto P, Hsiung GY, et al. (2012) Early-onset familial Alzheimer's disease (EOFAD). Can J Neurol Sci 39: 436-445. doi: 10.1017/S0317167100013949
|
| [25] |
Ballard C, Gauthier S, Corbett A, et al. (2011) Alzheimer's disease. Lancet 377: 1019-1031. doi: 10.1016/S0140-6736(10)61349-9
|
| [26] |
Castello MA, Jeppson JD, Soriano S (2014) Moving beyond anti-amyloid therapy for the prevention and treatment of Alzheimer’s disease. BMC Neurol 14: 1-5. doi: 10.1186/1471-2377-14-1
|
| [27] |
Cedazo-Minguez A, Winblad B (2010) Biomarkers for Alzheimer's disease and other forms of dementia: clinical needs, limitations and future aspects. Exp Gerontol 45: 5-14. doi: 10.1016/j.exger.2009.09.008
|
| [28] |
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci 12: 383-388. doi: 10.1016/0165-6147(91)90609-V
|
| [29] |
Puzzo D, Gulisano W, Arancio O, et al. (2015) The keystone of Alzheimer pathogenesis might be sought in Abeta physiology. Neuroscience 307: 26-36. doi: 10.1016/j.neuroscience.2015.08.039
|
| [30] |
Fiandaca MS, Mapstone ME, Cheema AK, et al. (2014) The critical need for defining preclinical biomarkers in Alzheimer'sdisease. Alzheimers Dement 10: S196-S212. doi: 10.1016/j.jalz.2014.04.015
|
| [31] |
Racchi M, Govoni S (2003) The pharmacology of amyloid precursor protein processing. Exp Gerontol 38: 145-157. doi: 10.1016/S0531-5565(02)00158-4
|
| [32] |
Nhan HS, Chiang K, Koo EH (2015) The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes. Acta Neuropathol 129: 1-19. doi: 10.1007/s00401-014-1347-2
|
| [33] |
Chasseigneaux S, Allinquant B (2012) Functions of Abeta, sAPPalpha and sAPPbeta : similarities and differences. J Neurochem 120 Suppl 1: 99-108. doi: 10.1111/j.1471-4159.2011.07584.x
|
| [34] | Simic G, Babic Leko M, Wray S, et al. (2016) Tau Protein Hyperphosphorylation and Aggregation in Alzheimer's Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 6. |
| [35] | Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17: 22-35. |
| [36] | Wang JZ, Xia YY, Grundke-Iqbal I, et al. (2013) Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis 33 Suppl 1: S123-139. |
| [37] |
Robakis NK (2010) Are Abeta and its derivatives causative agents or innocent bystanders in AD? Neurodegener Dis 7: 32-37. doi: 10.1159/000266476
|
| [38] |
Castello MA, Soriano S (2014) On the origin of Alzheimer's disease. Trials and tribulations of the amyloid hypothesis. Ageing Res Rev 13: 10-12. doi: 10.1016/j.arr.2013.10.001
|
| [39] |
Loy CT, Schofield PR, Turner AM, et al. (2014) Genetics of dementia. Lancet 383: 828-840. doi: 10.1016/S0140-6736(13)60630-3
|
| [40] |
Arriagada PV, Growdon JH, Hedley-Whyte ET, et al. (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42: 631-639. doi: 10.1212/WNL.42.3.631
|
| [41] |
Crystal H, Dickson D, Fuld P, et al. (1988) Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer's disease. Neurology 38: 1682-1687. doi: 10.1212/WNL.38.11.1682
|
| [42] |
Benilova I, Karran E, De Strooper B (2012) The toxic A[beta] oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci 15: 349-357. doi: 10.1038/nn.3028
|
| [43] |
Hardy J (2009) The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem 110: 1129-1134. doi: 10.1111/j.1471-4159.2009.06181.x
|
| [44] |
Hsia AY, Masliah E, McConlogue L, et al. (1999) Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A 96: 3228-3233. doi: 10.1073/pnas.96.6.3228
|
| [45] | Mucke L, Masliah E, Yu GQ, et al. (2000) High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20: 4050-4058. |
| [46] |
Holmes C, Boche D, Wilkinson D, et al. (2008) Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372: 216-223. doi: 10.1016/S0140-6736(08)61075-2
|
| [47] |
Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10: 698-712. doi: 10.1038/nrd3505
|
| [48] |
Robakis NK (2011) Mechanisms of AD neurodegeneration may be independent of Abeta and its derivatives. Neurobiol Aging 32: 372-379. doi: 10.1016/j.neurobiolaging.2010.05.022
|
| [49] |
Richens JL, Morgan K, O'Shea P (2014) Reverse engineering of Alzheimer's disease based on biomarker pathways analysis. Neurobiol Aging 35: 2029-2038. doi: 10.1016/j.neurobiolaging.2014.02.024
|
| [50] |
Morgan K (2011) The three new pathways leading to Alzheimer's disease. Neuropathol Appl Neurobiol 37: 353-357. doi: 10.1111/j.1365-2990.2011.01181.x
|
| [51] |
Baumgart M, Snyder HM, Carrillo MC, et al. (2015) Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimers Dement 11: 718-726. doi: 10.1016/j.jalz.2015.05.016
|
| [52] |
Hebert LE, Scherr PA, Bienias JL, et al. (2003) Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 60: 1119-1122. doi: 10.1001/archneur.60.8.1119
|
| [53] | Moser VA, Pike CJ (2015) Obesity and sex interact in the regulation of Alzheimer's disease. Neurosci Biobehav Rev in press. |
| [54] |
Li R, Singh M (2014) Sex differences in cognitive impairment and Alzheimer's disease. Front Neuroendocrinol 35: 385-403. doi: 10.1016/j.yfrne.2014.01.002
|
| [55] | Barron AM, Pike CJ (2012) Sex hormones, aging, and Alzheimer's disease. Front Biosci (Elite Ed) 4: 976-997. |
| [56] |
Luchsinger JA, Cheng D, Tang MX, et al. (2012) Central obesity in the elderly is related to late-onset Alzheimer disease. Alzheimer Dis Assoc Disord 26: 101-105. doi: 10.1097/WAD.0b013e318222f0d4
|
| [57] | Emmerzaal TL, Kiliaan AJ, Gustafson DR (2015) 2003-2013: a decade of body mass index, Alzheimer's disease, and dementia. J Alzheimers Dis 43: 739-755. |
| [58] |
Tsoi KK, Chan JY, Hirai HW, et al. (2015) Cognitive Tests to Detect Dementia: A Systematic Review and Meta-analysis. JAMA Intern Med 175: 1450-1458. doi: 10.1001/jamainternmed.2015.2152
|
| [59] |
Folstein MF, Folstein SE, McHugh PR (1975) "Mini-mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: 189-198. doi: 10.1016/0022-3956(75)90026-6
|
| [60] |
Mioshi E, Dawson K, Mitchell J, et al. (2006) The Addenbrooke's Cognitive Examination Revised (ACE-R): a brief cognitive test battery for dementia screening. Int J Geriatr Psychiatry 21: 1078-1085. doi: 10.1002/gps.1610
|
| [61] |
Larner AJ, Mitchell AJ (2014) A meta-analysis of the accuracy of the Addenbrooke's Cognitive Examination (ACE) and the Addenbrooke's Cognitive Examination-Revised (ACE-R) in the detection of dementia. Int Psychogeriatr 26: 555-563. doi: 10.1017/S1041610213002329
|
| [62] |
Hampel H, Frank R, Broich K, et al. (2010) Biomarkers for Alzheimer's disease: academic, industry and regulatory perspectives. Nat Rev Drug Discov 9: 560-574. doi: 10.1038/nrd3115
|
| [63] |
Corder EH, Saunders AM, Strittmatter WJ, et al. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921-923. doi: 10.1126/science.8346443
|
| [64] |
Saunders AM, Strittmatter WJ, Schmechel D, et al. (1993) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology 43: 1467-1472. doi: 10.1212/WNL.43.8.1467
|
| [65] | Xiang Y, Lam SM, Shui G (2015) What can lipidomics tell us about the pathogenesis of Alzheimer disease? Biol Chem 396: 1281-1291. |
| [66] |
Bertram L, McQueen MB, Mullin K, et al. (2007) Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39: 17-23. doi: 10.1038/ng1934
|
| [67] |
Corder EH, Saunders AM, Risch NJ, et al. (1994) Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 7: 180-184. doi: 10.1038/ng0694-180
|
| [68] |
Myers RH, Schaefer EJ, Wilson PW, et al. (1996) Apolipoprotein E epsilon4 association with dementia in a population-based study: The Framingham study. Neurology 46: 673-677. doi: 10.1212/WNL.46.3.673
|
| [69] |
Barber RC, Phillips NR, Tilson JL, et al. (2015) Can Genetic Analysis of Putative Blood Alzheimer's Disease Biomarkers Lead to Identification of Susceptibility Loci? PLoS One 10: e0142360. doi: 10.1371/journal.pone.0142360
|
| [70] | Van Cauwenberghe C, Van Broeckhoven C, Sleegers K (2015) The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med in press. |
| [71] |
Karch CM, Goate AM (2015) Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 77: 43-51. doi: 10.1016/j.biopsych.2014.05.006
|
| [72] |
Bertram L, Lill CM, Tanzi RE (2010) The genetics of Alzheimer disease: back to the future. Neuron 68: 270-281. doi: 10.1016/j.neuron.2010.10.013
|
| [73] |
Guerreiro R, Wojtas A, Bras J, et al. (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368: 117-127. doi: 10.1056/NEJMoa1211851
|
| [74] |
Harold D, Abraham R, Hollingworth P, et al. (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 41: 1088-1093. doi: 10.1038/ng.440
|
| [75] |
Hollingworth P, Harold D, Sims R, et al. (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 43: 429-435. doi: 10.1038/ng.803
|
| [76] |
Lambert JC, Heath S, Even G, et al. (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41: 1094-1099. doi: 10.1038/ng.439
|
| [77] |
Naj AC, Jun G, Beecham GW, et al. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 43: 436-441. doi: 10.1038/ng.801
|
| [78] |
Seshadri S, Fitzpatrick AL, Ikram MA, et al. (2010) Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303: 1832-1840. doi: 10.1001/jama.2010.574
|
| [79] |
Huang Y (2010) Abeta-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer's disease. Trends Mol Med 16: 287-294. doi: 10.1016/j.molmed.2010.04.004
|
| [80] |
Kim J, Yoon H, Basak J, et al. (2014) Apolipoprotein E in synaptic plasticity and Alzheimer's disease: potential cellular and molecular mechanisms. Mol Cells 37: 767-776. doi: 10.14348/molcells.2014.0248
|
| [81] |
DeMattos RB, Cirrito JR, Parsadanian M, et al. (2004) ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron 41: 193-202. doi: 10.1016/S0896-6273(03)00850-X
|
| [82] |
Nathan BP, Bellosta S, Sanan DA, et al. (1994) Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science 264: 850-852. doi: 10.1126/science.8171342
|
| [83] |
Ji Y, Gong Y, Gan W, et al. (2003) Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer's disease patients. Neuroscience 122: 305-315. doi: 10.1016/j.neuroscience.2003.08.007
|
| [84] |
Lidstrom AM, Bogdanovic N, Hesse C, et al. (1998) Clusterin (apolipoprotein J) protein levels are increased in hippocampus and in frontal cortex in Alzheimer's disease. Exp Neurol 154: 511-521. doi: 10.1006/exnr.1998.6892
|
| [85] |
Sihlbom C, Davidsson P, Sjogren M, et al. (2008) Structural and quantitative comparison of cerebrospinal fluid glycoproteins in Alzheimer's disease patients and healthy individuals. Neurochem Res 33: 1332-1340. doi: 10.1007/s11064-008-9588-x
|
| [86] |
Nilselid AM, Davidsson P, Nagga K, et al. (2006) Clusterin in cerebrospinal fluid: analysis of carbohydrates and quantification of native and glycosylated forms. Neurochem Int 48: 718-728. doi: 10.1016/j.neuint.2005.12.005
|
| [87] |
Jongbloed W, van Dijk KD, Mulder SD, et al. (2015) Clusterin Levels in Plasma Predict Cognitive Decline and Progression to Alzheimer's Disease. J Alzheimers Dis 46: 1103-1110. doi: 10.3233/JAD-150036
|
| [88] |
Thambisetty M, Simmons A, Velayudhan L, et al. (2010) Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry 67: 739-748. doi: 10.1001/archgenpsychiatry.2010.78
|
| [89] |
Schrijvers EM, Koudstaal PJ, Hofman A, et al. (2011) Plasma clusterin and the risk of Alzheimer disease. JAMA 305: 1322-1326. doi: 10.1001/jama.2011.381
|
| [90] |
McGeer PL, Kawamata T, Walker DG (1992) Distribution of clusterin in Alzheimer brain tissue. Brain Res 579: 337-341. doi: 10.1016/0006-8993(92)90071-G
|
| [91] | Ghiso J, Matsubara E, Koudinov A, et al. (1993) The cerebrospinal-fluid soluble form of Alzheimer's amyloid beta is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochem J 293 (Pt 1): 27-30. |
| [92] |
Zlokovic BV (1996) Cerebrovascular transport of Alzheimer's amyloid beta and apolipoproteins J and E: possible anti-amyloidogenic role of the blood-brain barrier. Life Sci 59: 1483-1497. doi: 10.1016/0024-3205(96)00310-4
|
| [93] |
Giannakopoulos P, Kovari E, French LE, et al. (1998) Possible neuroprotective role of clusterin in Alzheimer's disease: a quantitative immunocytochemical study. Acta Neuropathol 95: 387-394. doi: 10.1007/s004010050815
|
| [94] |
Oda T, Wals P, Osterburg HH, et al. (1995) Clusterin (apoJ) alters the aggregation of amyloid beta-peptide (A beta 1-42) and forms slowly sedimenting A beta complexes that cause oxidative stress. Exp Neurol 136: 22-31. doi: 10.1006/exnr.1995.1080
|
| [95] |
Li X, Ma Y, Wei X, et al. (2014) Clusterin in Alzheimer's disease: a player in the biological behavior of amyloid-beta. Neurosci Bull 30: 162-168. doi: 10.1007/s12264-013-1391-2
|
| [96] |
Jiang T, Tan L, Zhu XC, et al. (2014) Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer's disease. Neuropsychopharmacology 39: 2949-2962. doi: 10.1038/npp.2014.164
|
| [97] |
Zhao Y, Bhattacharjee S, Jones BM, et al. (2013) Regulation of TREM2 expression by an NF-small ka, CyrillicB-sensitive miRNA-34a. Neuroreport 24: 318-323. doi: 10.1097/WNR.0b013e32835fb6b0
|
| [98] |
Lue LF, Schmitz C, Walker DG (2015) What happens to microglial TREM2 in Alzheimer's disease: Immunoregulatory turned into immunopathogenic? Neuroscience 302: 138-150. doi: 10.1016/j.neuroscience.2014.09.050
|
| [99] |
Jiang T, Yu JT, Zhu XC, et al. (2013) TREM2 in Alzheimer's disease. Mol Neurobiol 48: 180-185. doi: 10.1007/s12035-013-8424-8
|
| [100] |
Frank S, Burbach GJ, Bonin M, et al. (2008) TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56: 1438-1447. doi: 10.1002/glia.20710
|
| [101] |
Paradowska-Gorycka A, Jurkowska M (2013) Structure, expression pattern and biological activity of molecular complex TREM-2/DAP12. Hum Immunol 74: 730-737. doi: 10.1016/j.humimm.2013.02.003
|
| [102] |
Paris D, Ait-Ghezala G, Bachmeier C, et al. (2014) The spleen tyrosine kinase (Syk) regulates Alzheimer amyloid-beta production and Tau hyperphosphorylation. J Biol Chem 289: 33927-33944. doi: 10.1074/jbc.M114.608091
|
| [103] |
Lebouvier T, Scales TM, Hanger DP, et al. (2008) The microtubule-associated protein tau is phosphorylated by Syk. Biochim Biophys Acta 1783: 188-192. doi: 10.1016/j.bbamcr.2007.11.005
|
| [104] | Vafadar-Isfahani B, Ball G, Coveney C, et al. (2012) Identification of SPARC-like 1 Protein as Part of a Biomarker Panel for Alzheimer's Disease in Cerebrospinal Fluid. J Alzheimers Dis 28: 625-636. |
| [105] | Richens JL, Vere KA, Light RA, et al. (2014) Practical detection of a definitive biomarker panel for Alzheimer's disease; comparisons between matched plasma and cerebrospinal fluid. Int J Mol Epidemiol Genet 5: 53-70. |
| [106] |
Mendis DB, Ivy GO, Brown IR (2000) Induction of SC1 mRNA encoding a brain extracellular matrix glycoprotein related to SPARC following lesioning of the adult rat forebrain. Neurochem Res 25: 1637-1644. doi: 10.1023/A:1026626805612
|
| [107] |
Yin GN, Lee HW, Cho JY, et al. (2009) Neuronal pentraxin receptor in cerebrospinal fluid as a potential biomarker for neurodegenerative diseases. Brain Res 1265: 158-170. doi: 10.1016/j.brainres.2009.01.058
|
| [108] |
Xu PT, Li YJ, Qin XJ, et al. (2006) Differences in apolipoprotein E3/3 and E4/4 allele-specific gene expression in hippocampus in Alzheimer disease. Neurobiol Dis 21: 256-275. doi: 10.1016/j.nbd.2005.07.004
|
| [109] | Abdi F, Quinn JF, Jankovic J, et al. (2006) Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neurodegenerative disorders. J Alzheimers Dis 9: 293-348. |
| [110] |
Weisel JW (2005) Fibrinogen and fibrin. Adv Protein Chem 70: 247-299. doi: 10.1016/S0065-3233(05)70008-5
|
| [111] | Cortes-Canteli M, Strickland S (2009) Fibrinogen, a possible key player in Alzheimer's disease. J Thromb Haemost 7 Suppl 1: 146-150. |
| [112] |
Cortes-Canteli M, Paul J, Norris EH, et al. (2010) Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer's disease. Neuron 66: 695-709. doi: 10.1016/j.neuron.2010.05.014
|
| [113] |
Ahn HJ, Glickman JF, Poon KL, et al. (2014) A novel Abeta-fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer's disease mice. J Exp Med 211: 1049-1062. doi: 10.1084/jem.20131751
|
| [114] |
Ahn HJ, Zamolodchikov D, Cortes-Canteli M, et al. (2010) Alzheimer's disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci U S A 107: 21812-21817. doi: 10.1073/pnas.1010373107
|
| [115] | Mueller C, Zhou W, Vanmeter A, et al. (2010) The heme degradation pathway is a promising serum biomarker source for the early detection of Alzheimer's disease. J Alzheimers Dis 19: 1081-1091. |
| [116] |
Langbein L, Heid HW, Moll I, et al. (1993) Molecular characterization of the body site-specific human epidermal cytokeratin 9: cDNA cloning, amino acid sequence, and tissue specificity of gene expression. Differentiation 55: 57-71. doi: 10.1111/j.1432-0436.1993.tb00033.x
|
| [117] | Ghosh D, Lippert D, Krokhin O, et al. (2010) Defining the membrane proteome of NK cells. J Mass Spectrom 45: 1-25. |
| [118] |
van Niel G, Raposo G, Candalh C, et al. (2001) Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology 121: 337-349. doi: 10.1053/gast.2001.26263
|
| [119] |
de Mateo S, Castillo J, Estanyol JM, et al. (2011) Proteomic characterization of the human sperm nucleus. Proteomics 11: 2714-2726. doi: 10.1002/pmic.201000799
|
| [120] | Bohm D, Keller K, Pieter J, et al. (2012) Comparison of tear protein levels in breast cancer patients and healthy controls using a de novo proteomic approach. Oncol Rep 28: 429-438. |
| [121] | Kim YS, Gu BH, Choi BC, et al. (2013) Apolipoprotein A-IV as a novel gene associated with polycystic ovary syndrome. Int J Mol Med 31: 707-716. |
| [122] | Fu BS, Liu W, Zhang JW, et al. (2009) [Serum proteomic analysis on metastasis-associated proteins of hepatocellular carcinoma]. Nan Fang Yi Ke Da Xue Xue Bao 29: 1775-1778. |
| [123] |
Li X, Long J, He T, et al. (2015) Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer's disease. Sci Rep 5: 12393. doi: 10.1038/srep12393
|
| [124] | Femminella GD, Ferrara N, Rengo G (2015) The emerging role of microRNAs in Alzheimer's disease. Front Physiol 6: 40. |
| [125] |
Lukiw WJ, Zhao Y, Cui JG (2008) An NF-kappaB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J Biol Chem 283: 31315-31322. doi: 10.1074/jbc.M805371200
|
| [126] |
Zhu HC, Wang LM, Wang M, et al. (2012) MicroRNA-195 downregulates Alzheimer's disease amyloid-beta production by targeting BACE1. Brain Res Bull 88: 596-601. doi: 10.1016/j.brainresbull.2012.05.018
|
| [127] |
Santa-Maria I, Alaniz ME, Renwick N, et al. (2015) Dysregulation of microRNA-219 promotes neurodegeneration through post-transcriptional regulation of tau. J Clin Invest 125: 681-686. doi: 10.1172/JCI78421
|
| [128] |
Kosicek M, Zetterberg H, Andreasen N, et al. (2012) Elevated cerebrospinal fluid sphingomyelin levels in prodromal Alzheimer's disease. Neurosci Lett 516: 302-305. doi: 10.1016/j.neulet.2012.04.019
|
| [129] |
Mulder C, Wahlund LO, Teerlink T, et al. (2003) Decreased lysophosphatidylcholine/phosphatidylcholine ratio in cerebrospinal fluid in Alzheimer's disease. J Neural Transm (Vienna) 110: 949-955. doi: 10.1007/s00702-003-0007-9
|
| [130] |
Satoi H, Tomimoto H, Ohtani R, et al. (2005) Astroglial expression of ceramide in Alzheimer's disease brains: a role during neuronal apoptosis. Neuroscience 130: 657-666. doi: 10.1016/j.neuroscience.2004.08.056
|
| [131] |
Mapstone M, Cheema AK, Fiandaca MS, et al. (2014) Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med 20: 415-418. doi: 10.1038/nm.3466
|
| [132] |
Wood PL, Medicherla S, Sheikh N, et al. (2015) Targeted Lipidomics of Fontal Cortex and Plasma Diacylglycerols (DAG) in Mild Cognitive Impairment and Alzheimer's Disease: Validation of DAG Accumulation Early in the Pathophysiology of Alzheimer's Disease. J Alzheimers Dis 48: 537-546. doi: 10.3233/JAD-150336
|
| [133] |
Hu Z, Chang YC, Wang Y, et al. (2013) VisANT 4.0: Integrative network platform to connect genes, drugs, diseases and therapies. Nucleic Acids Res 41: W225-231. doi: 10.1093/nar/gkt401
|
| [134] |
Chatr-Aryamontri A, Breitkreutz BJ, Oughtred R, et al. (2015) The BioGRID interaction database: 2015 update. Nucleic Acids Res 43: D470-478. doi: 10.1093/nar/gku1204
|
| [135] | Johnson C, Tinti M, Wood NT, et al. (2011) Visualization and biochemical analyses of the emerging mammalian 14-3-3-phosphoproteome. Mol Cell Proteomics 10: M110 005751. |
| [136] | Reddy A, Huang CC, Liu H, et al. (2010) Robust gene network analysis reveals alteration of the STAT5a network as a hallmark of prostate cancer. Genome Inform 24: 139-153. |
| [137] |
Araujo DJ, Anderson AG, Berto S, et al. (2015) FoxP1 orchestration of ASD-relevant signaling pathways in the striatum. Genes Dev 29: 2081-2096. doi: 10.1101/gad.267989.115
|
| [138] | Haas LT, Salazar SV, Kostylev MA, et al. (2015) Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer's disease. Brain 139: 526-546. |
| [139] |
Shahani N, Seshadri S, Jaaro-Peled H, et al. (2015) DISC1 regulates trafficking and processing of APP and Abeta generation. Mol Psychiatry 20: 874-879. doi: 10.1038/mp.2014.100
|
| [140] |
Croft D, O'Kelly G, Wu G, et al. (2011) Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res 39: D691-697. doi: 10.1093/nar/gkq1018
|
| [141] |
D'Eustachio P (2013) Pathway Databases: Making Chemical and Biological Sense of the Genomic Data Flood. Chem Biol 20: 629-635. doi: 10.1016/j.chembiol.2013.03.018
|
| [142] |
Wang X, Liotta L (2011) Clinical bioinformatics: a new emerging science. J Clin Bioinforma 1: 1. doi: 10.1186/2043-9113-1-1
|
| [143] |
Bellazzi R, Masseroli M, Murphy S, et al. (2012) Clinical Bioinformatics: challenges and opportunities. BMC Bioinform 13: 1-8. doi: 10.1186/1471-2105-13-1
|
| [144] |
Butte AJ (2008) Translational Bioinformatics: Coming of Age. J Am Med Inf Assoc 15: 709-714. doi: 10.1197/jamia.M2824
|
| [145] |
Larrañaga P, Calvo B, Santana R, et al. (2006) Machine learning in bioinformatics. Brief Bioinform 7: 86-112. doi: 10.1093/bib/bbk007
|
| [146] |
Pavlopoulos GA, Secrier M, Moschopoulos CN, et al. (2011) Using graph theory to analyze biological networks. BioData Min 4: 1-27. doi: 10.1186/1756-0381-4-1
|
| [147] |
Bullmore E, Sporns O (2009) Complex brain networks: graph theoretical analysis of structural and functional systems. Nat Rev Neurosci 10: 186-198. doi: 10.1038/nrn2575
|
| [148] |
Stam CJ (2014) Modern network science of neurological disorders. Nat Rev Neurosci 15: 683-695. doi: 10.1038/nrn3801
|
| [149] |
Baumgartner C, Osl M, Netzer M, et al. (2011) Bioinformatic-driven search for metabolic biomarkers in disease. J Clin Bioinforma 1: 1-10. doi: 10.1186/2043-9113-1-1
|
| [150] | Inza I, Calvo B, Armañanzas R, et al. (2010) Machine Learning: An Indispensable Tool in Bioinformatics. In: Matthiesen R, editor. Bioinformatics Methods in Clinical Research: Humana Press. pp. 25-48. |
| [151] | Guyon I, Weston J, Barnhill S, et al. Gene Selection for Cancer Classification using Support Vector Machines. Mach Learn 46: 389-422. |
| [152] | Hsu W-C, Denq C, Chen S-S (2013) A diagnostic methodology for Alzheimer’s disease. J Clin Bioinforma 3: 1-13. |
| [153] |
Horwitz B, Rowe JB (2011) Functional biomarkers for neurodegenerative disorders based on the network paradigm. Prog Neurobiol 95: 505-509. doi: 10.1016/j.pneurobio.2011.07.005
|
| [154] |
de Haan W, Pijnenburg YAL, Strijers RLM, et al. (2009) Functional neural network analysis in frontotemporal dementia and Alzheimer's disease using EEG and graph theory. BMC Neurosci 10: 101-101. doi: 10.1186/1471-2202-10-101
|
| [155] |
Brier MR, Thomas JB, Fagan AM, et al. (2014) Functional connectivity and graph theory in preclinical Alzheimer's disease. Neurobiol Aging 35: 757-768. doi: 10.1016/j.neurobiolaging.2013.10.081
|
| [156] |
Zhang D, Wang Y, Zhou L, et al. (2011) Multimodal classification of Alzheimer's disease and mild cognitive impairment. NeuroImage 55: 856-867. doi: 10.1016/j.neuroimage.2011.01.008
|
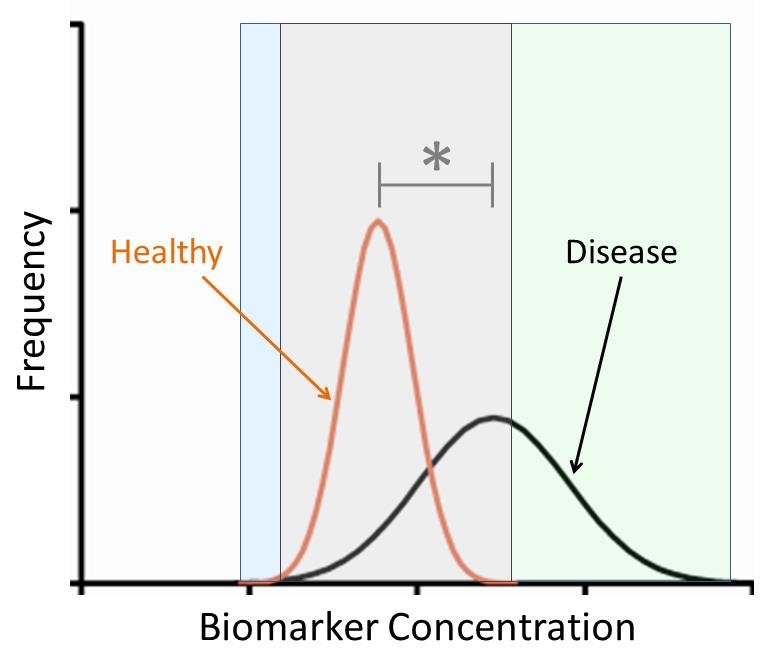
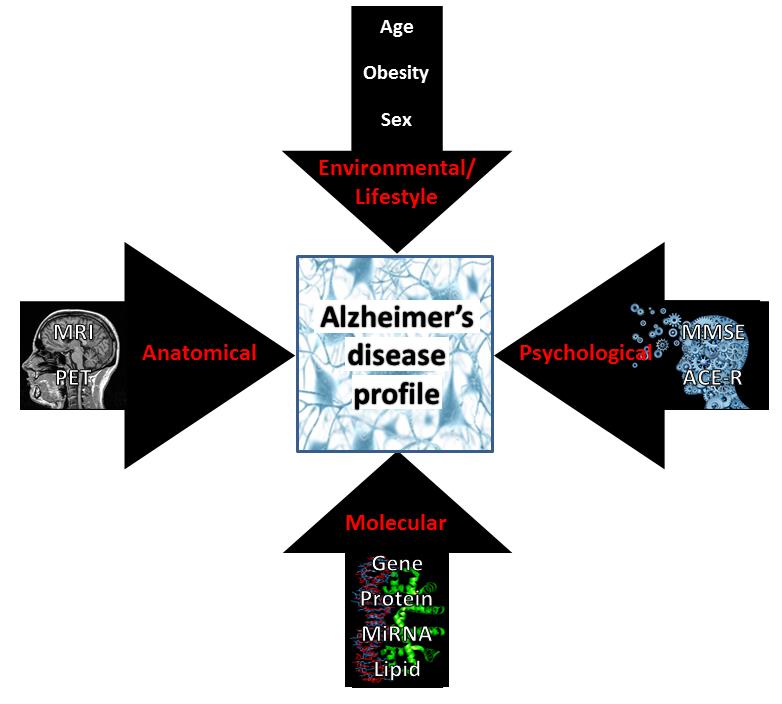
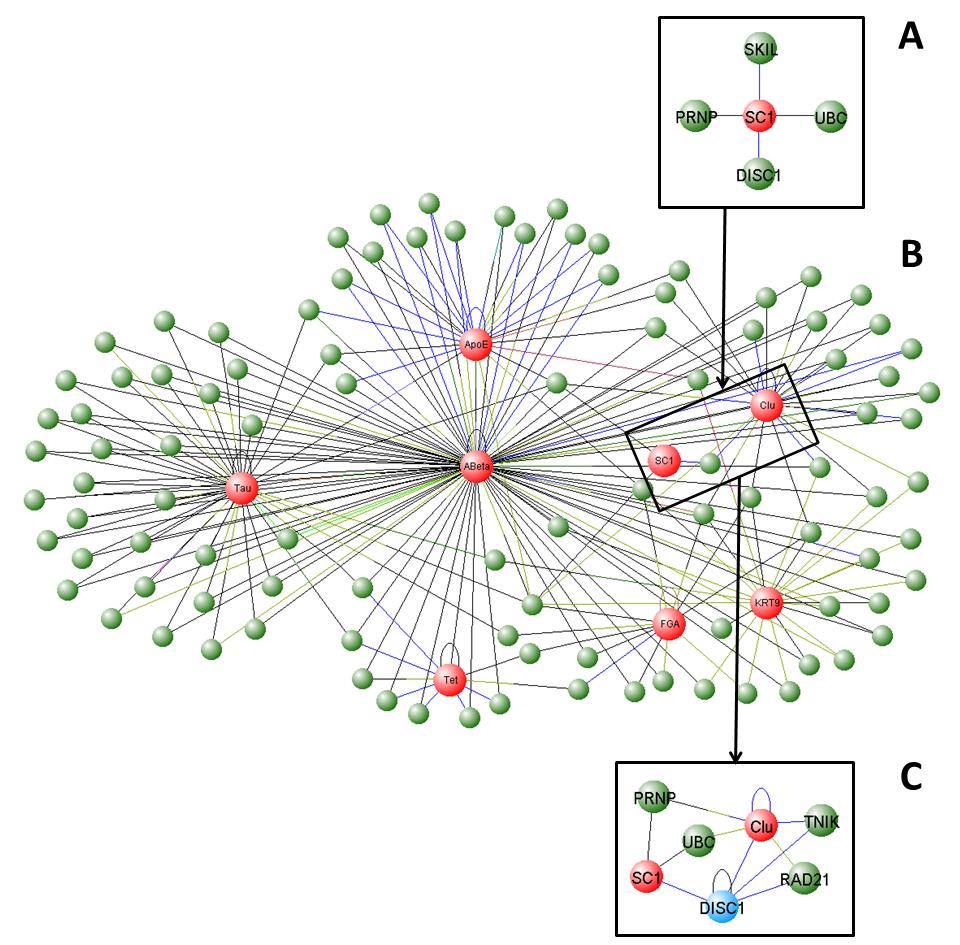
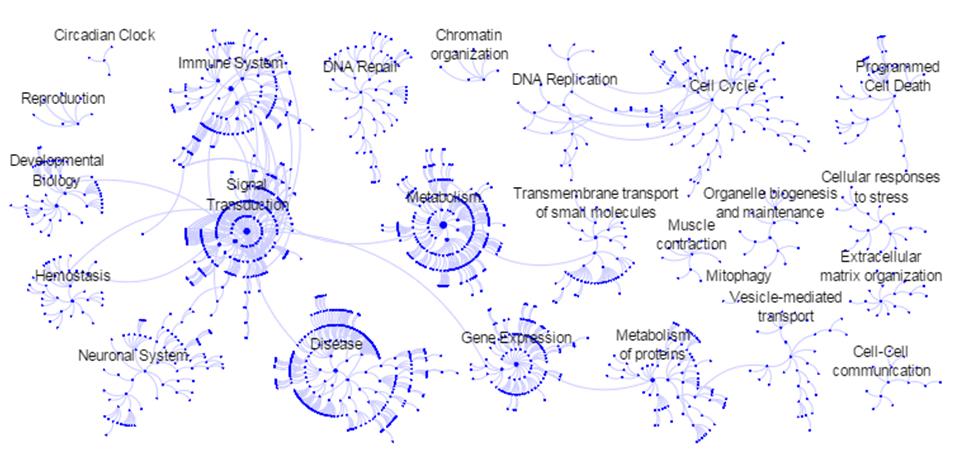
Figures(5)

Joanna L. Richens, Jonathan P. Bramble, Hannah L. Spencer, Fiona Cantlay, Molly Butler, Paul O’Shea. Towards defining the Mechanisms of Alzheimer’s disease based on a contextual analysis of molecular pathways[J]. AIMS Genetics, 2016, 3(1): 25-48. doi: 10.3934/genet.2016.1.25







 DownLoad:
DownLoad: