Citation: Marina A. Darenskaya, Olga V. Smirnova, Boris G. Gubanov, Natalya V. Semenova, Lyubov I. Kolesnikova, Sergey I. Kolesnikov. Lipid peroxidation, antioxidant defense parameters, and dynamics of surgical treatment in men with mechanical jaundice of various origins[J]. AIMS Molecular Science, 2020, 7(4): 374-382. doi: 10.3934/molsci.2020018

| [1] | Khan RS, Houlihan DD, Newsome PN (2019) Investigation of jaundice. Medicine 47: 713-717. |
| [2] | Zakaria AS, Gamil M, Ali Eldin NH, et al. (2019) P5: Malignant obstructive jaundice; review of 232 Patients. Pan Arab J Oncol 12: 55. |
| [3] | Pavlidis ET, Pavlidis TE (2018) Pathophysiological consequences of obstructive jaundice and perioperative management. Hepatobiliary Pancreatic Dis Int 17: 17-21. |
| [4] | Papadopoulos V, Filippou D, Manolis E, et al. (2007) Haemostasis impairment in patients with obstructive jaundice. J Gastrointestin Liver Dis 16: 177. |
| [5] | Huynh F, Usatoff V (2019) Obstructive jaundice. Textbook of Surgery New York: John Wiley & Sons, Ltd, 643-647. |
| [6] | Shevchenko BF, Zeleniuk OV, Klenina IA, et al. (2019) Structural and functional state of the liver in patients with extrahepatic cholestasis of non-tumor genesis. Rep Morphol 25: 36-43. |
| [7] | Hajiyev JM, Taghiyev EG, Hajiyev NJ (2016) Comparative evaluation of interleukin-6 in the liver tissues, bile duct, blood serum and urine in patients with obstructive jaundice of benign etiology. J Exp Clin Surg 9: 33-38. |
| [8] | Martínez-Cecilia D, Reyes-Díaz M, Ruiz-Rabelo J, et al. (2016) Oxidative stress influence on renal dysfunction in patients with obstructive jaundice: A case and control prospective study. Redox Biol 8: 160-164. |
| [9] | Moole H, Bechtold M, Puli SR (2016) Efficacy of preoperative biliary drainage in malignant obstructive jaundice: a meta-analysis and systematic review. World J Surg Oncol 14: 182. |
| [10] | Luo W (2017) Research advances in animal models of obstructive jaundice. J Clin Hepatol 33: 1820-1823. |
| [11] | Wang N, Han Q, Wang G, et al. (2016) Resveratrol protects oxidative stress-induced intestinal epithelial barrier dysfunction by upregulating heme oxygenase-1 expression. Dig Dis Sci 61: 2522-2534. |
| [12] | Tian X, Zhao H, Zhang Z, et al. (2019) Intestinal mucosal injury induced by obstructive jaundice is associated with activation of TLR4/TRAF6/NF-κB pathways. PLoS One 14: e0223651. |
| [13] | Assimakopoulos SF, Vagianos CE, Patsoukis N, et al. (2004) Evidence for intestinal oxidative stress in obstructive jaundice-induced gut barrier dysfunction in rats. Acta Physiol Scand 180: 177-185. |
| [14] | Assimakopoulos SF, Scopa CD, Vagianos CE (2007) Pathophysiology of increased intestinal permeability in obstructive jaundice. World J Gastroenterol 13: 6458. |
| [15] | Corongiu FP, Banni S (1994) Detection of conjugated dienes by second derivative ultraviolet spectrophotometry. Methods Enzymol 23: 303-310. |
| [16] | Asakava T, Matsushita S (1980) Coloring conditions of thiobarbituric acid test, for detecting lipid hydroperoxides. Lipids 15: 137-140. |
| [17] | Colowick SP, Kaplan NO (1984) Methods in Enzymology Orlando: Academic Press. |
| [18] | Misra HP, Fridovich I (1972) The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem 247: 3170-3175. |
| [19] | Schosinsky KH, Lehmann HP, Beeler MF (1974) Measurement of ceruloplasmin from its oxidase activity in serum by use of o-dianisidine dihydrochloride. Clin Chem 20: 1556-1563. |
| [20] | Rani V, Deep G, Singh RK, et al. (2016) Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci 148: 183-193. |
| [21] | Kolesnikova LI, Darenskaya MA, Kolesnikov SI (2017) Free radical oxidation: a pathophysiologist's view. Bull Sib Med 16: 16-29. |
| [22] | Kolesnikova LI, Semyonova NV, Grebenkina LA, et al. (2014) Integral indicator of oxidative stress in human blood. Bull Exp Biol Med 157: 715-717. |
| [23] | Vagholkar K (2020) Obstructive jaundice: understanding the pathophysiology. Int J Surg Med 6: 26-31. |
| [24] | Jing LI, Wu GL, Niu Q, et al. (2018) Bacterial culture of bile from patients with obstructive jaundice and antibiotic sensitivity. China J Endoscopy 24: 94-99. |
| [25] | Yang R, Zhu S, Pischke SE, et al. (2018) Bile and circulating HMGB1 contributes to systemic inflammation in obstructive jaundice. J Surg Res 228: 14-19. |
| [26] | Chowdhury AH, Camara M, Martinez-Pomares L, et al. (2016) Immune dysfunction in patients with obstructive jaundice before and after endoscopic retrograde cholangiopancreatography. Clin Sci 130: 1535-1544. |
| [27] | Sharifi-Rad M, Anil Kumar NV, Zucca P, et al. (2020) Lifestyle, oxidative stress, and antioxidants: Back and forth in the pathophysiology of chronic diseases. Front Physiol 11: 694. |
| [28] | Bjørklund G, Chirumbolo S (2017) Role of oxidative stress and antioxidants in daily nutrition and human health. Nutrition 33: 311-321. |
| [29] | Calabrese G, Morgan B, Riemer J (2017) Mitochondrial glutathione: regulation and functions. Antioxid Redox Signaling 27: 1162-1177. |
| [30] | Linder MC (2016) Ceruloplasmin and other copper binding components of blood plasma and their functions: an update. Metallomics 8: 887-905. |
| [31] | Onalan AK, Tuncal S, Kilicoglu S, et al. (2016) Effect of silymarin on oxidative stress and liver histopathology in experimental obstructive jaundice model. Acta Cir Bras 31: 801-806. |
| [32] | Savdan M, Çakir M, Vatansev H, et al. (2017) Preventing oxygen free radical damage by proanthocyanidin in obstructive jaundice. Turk J Surg 33: 62. |
| [33] | Winterbourn CC, Kettle AJ, Hampton MB (2016) Reactive oxygen species and neutrophil function. Annu Rev Biochem 85: 765-792. |
| [34] | Scheufele F, Schorn S, Demir IE, et al. (2017) Preoperative biliary stenting versus operation first in jaundiced patients due to malignant lesions in the pancreatic head: A meta-analysis of current literature. Surgery 161: 939-950. |
| [35] | Chang ZK, Kou ZP, Li SX, et al. (2018) To evaluate the correlation between the change of immune system function before and after the treatment of malignant obstructive type jaundice treated with biliary stent. Eur Rev Med Pharmacol Sci 22: 1638-1644. |
| [36] | Nehéz L, Andersson R (2002) Compromise of immune function in obstructive jaundice. Eur J Surg 168: 315-328. |
| [37] | Mehta MM, Weinberg SE, Chandel NS (2017) Mitochondrial control of immunity: beyond ATP. Nat Rev Immunol 17: 608-620. |
| [38] | Negre-Salvayre A, Auge N, Ayala V, et al. (2010) Pathological aspects of lipid peroxidation. Free Radical Res 44: 1125-1171. |
| [39] | Vieira SA, Zhang G, Decker EA (2017) Biological implications of lipid oxidation products. J Am Oil Chem Soc 94: 339-351. |

Figures(2)

Marina A. Darenskaya, Olga V. Smirnova, Boris G. Gubanov, Natalya V. Semenova, Lyubov I. Kolesnikova, Sergey I. Kolesnikov. Lipid peroxidation, antioxidant defense parameters, and dynamics of surgical treatment in men with mechanical jaundice of various origins[J]. AIMS Molecular Science, 2020, 7(4): 374-382. doi: 10.3934/molsci.2020018

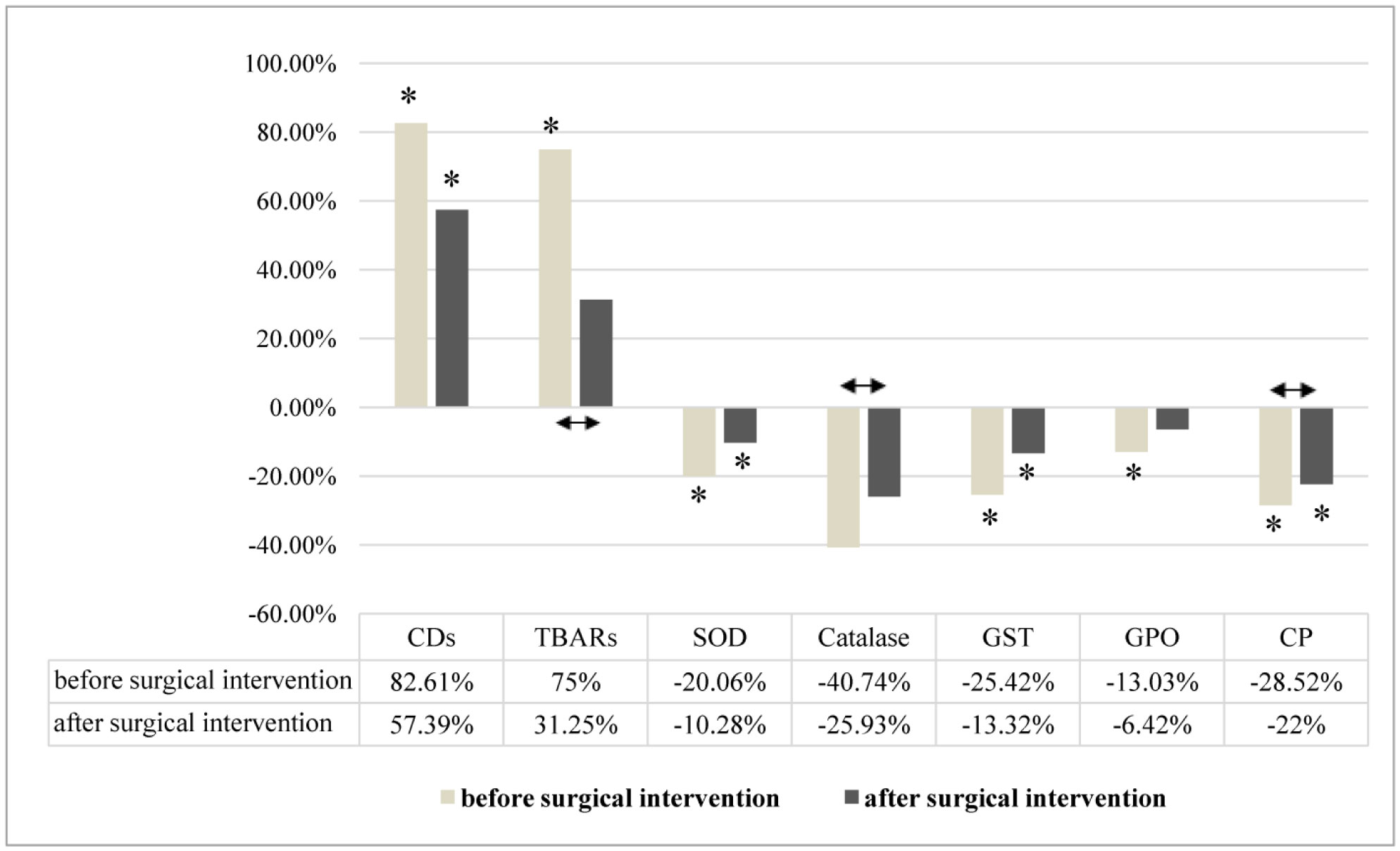
 indicates statistically significant differences in men with MJNT after surgical intervention compared to group before surgical intervention
indicates statistically significant differences in men with MJT after surgical intervention compared to group before surgical intervention
indicates statistically significant differences in men with MJNT after surgical intervention compared to group before surgical intervention
indicates statistically significant differences in men with MJT after surgical intervention compared to group before surgical intervention






 DownLoad:
DownLoad: