Citation: Ramakrishnan Ramugounder. The impact of p38 MAPK, 5-HT/DA/E signaling pathways in the development and progression of cardiovascular diseases and heart failure in type 1 diabetes[J]. AIMS Molecular Science, 2020, 7(4): 349-373. doi: 10.3934/molsci.2020017

| [1] |
Ramakrishnan R, Namasivayam A (1995) Norepinephrine and epinephrine levels in the brain of alloxan diabetic rats. Neurosci Lett 186: 200-202. doi: 10.1016/0304-3940(95)11315-N

|
| [2] | Ramakrishnan R, Suthanthirarajan N, Namasivayam A (1996) Brain dopamine in experimental diabetes. Indian J Physiol Pharmacol 40: 193-195. |
| [3] |
Ramakrishnan R, Nazer MY, Suthanthirarajan N, et al. (2003) An experimental analysis of the catecholamine's in hyperglycemia and acidosis induced rat brain. Int J Immunopathol Pharmacol 16: 233-239. doi: 10.1177/039463200301600308
|
| [4] |
Ramakrishnan R, Kempuraj D, Prabhakaran K, et al. (2005) A short-term diabetes induced changes of catecholamine's and p38 MAPK in discrete areas of rat brain. Life Sci 77: 1825-1835. doi: 10.1016/j.lfs.2004.12.038
|
| [5] | Ramakrishnan R (2014) Brain Biogenic Amines in Diabetes LAP Lambert Academic Publishing, 1-148. |
| [6] |
Ramakrishnan R (2019) Brain signaling systems: A target for treating type 1 diabetes mellitus. Brain Res Bull 152: 191-201. doi: 10.1016/j.brainresbull.2019.07.017
|
| [7] |
Shpakov AO, Derkach KV, Berstein LM (2015) Brain signaling systems in the Type 2 diabetes and metabolic syndrome: promising target to treat and prevent these diseases. Future Sci OA 1: FSO25. doi: 10.4155/fso.15.23
|
| [8] |
Watson AMD, Gould EAM, Penfold SA, et al. (2019) Diabetes and Hypertension Differentially Affect Renal Catecholamines and Renal Reactive Oxygen Species. Front Physiol 10: 309. doi: 10.3389/fphys.2019.00309
|
| [9] | Akalu Y, Birhan A (2020) Peripheral arterial disease and its associated factors among type 2 diabetes mellitus patients at Debre Tabor general hospital, Northwest Ethiopia. J Diabetes Res 9419413. |
| [10] |
Ramakrishnan R, Sheeladevi R, Suthanthirarajan N (2004) PKC-alpha mediated alterations of indoleamine contents in diabetic rat brain. Brain Res Bull 64: 189-194. doi: 10.1016/j.brainresbull.2004.07.002
|
| [11] |
Ramakrishnan R, Prabhakaran K, Jayakumar AR, et al. (2005) Involvement of Ca(2+)/calmodulin-dependent protein kinase II in the modulation of indolamines in diabetic and hyperglycemic rats. J Neurosci Res 80: 518-528. doi: 10.1002/jnr.20499
|
| [12] |
Ramakrishnan R, Sheeladevi R, Suthanthirarajan N, et al. (2005) An acute hyperglycemia or acidosis-induced changes of indolamines level correlates with PKC-alpha expression in rat brain. Brain Res Bull 67: 46-52. doi: 10.1016/j.brainresbull.2005.06.001
|
| [13] |
Ramakrishnan R, Sheeladevi R, Namasivayam A (2009) Regulation of protein kinases and co-regulatory interplay of S-100β between PKAII and PKC-α on serotonin level in diabetic rat brain. J Neurosci Res 87: 246-259. doi: 10.1002/jnr.21833
|
| [14] |
Moran C, Phan TG, Chen J, et al. (2013) Brain Atrophy in Type 2 Diabetes: Regional distribution and influence on cognition. Diabetes Care 36: 4036-4042. doi: 10.2337/dc13-0143
|
| [15] |
Moran C, Beare R, Wang W, et al. (2019) Type 2 diabetes mellitus, brain atropy, and cognitive decline. Neurology 92. doi: 10.1212/WNL.0000000000006955
|
| [16] |
Chen K (2004) Organization of MAO A and MAO B promoters and regulation of gene expression. Neurotoxicology 25: 31-36. doi: 10.1016/S0161-813X(03)00113-X
|
| [17] |
Fang C, Wu B, Le NTT, et al. (2018) Prions activate a p38 MAPK synaptotoxic signaling pathway. PLoS Pathog 14: e1007283. doi: 10.1371/journal.ppat.1007283
|
| [18] | Dewi DAMS, Wiryana M (2019) The interaction of neuroimmunology, neuromodulator, and neurotransmitter with nociceptor and MAPK signaling. J Immunol Res Ther 4: 9. |
| [19] | Yang X, Guo Z, Lu J, et al. (2017) The Role of MAPK and Dopaminergic Synapse Signaling Pathways in Antidepressant Effect of Electroacupuncture Pretreatment in Chronic Restraint Stress Rats. Evid Based Complement Alternat Med 2357653. |
| [20] |
Elliott G, Juan BG, Jacqueline HF, et al. (2017) Serotonin and catecholamine's in the development and progression of heart valve diseases. Cardiovasc Res 113: 849-857. doi: 10.1093/cvr/cvx092
|
| [21] |
Rosano GMC, Vitale C, Seferovic P (2017) Heart Failure in Patients with Diabetes Mellitus. Card Fail Rev 3: 52-55. doi: 10.15420/cfr.2016:20:2
|
| [22] |
Packer M (2018) Heart Failure: The Most Important, Preventable, and Treatable Cardiovascular Complication of Type 2 Diabetes. Diabetes Care 41: 11-13. doi: 10.2337/dci17-0052
|
| [23] |
Kenny HC, Abel ED (2019) Heart Failure in Type 2 Diabetes Mellitus: Impact of Glucose-Lowering Agents, Heart Failure Therapies, and Novel Therapeutic Strategies. Circ Res 124: 121-141. doi: 10.1161/CIRCRESAHA.118.311371
|
| [24] |
De Vecchis R, Cantatrione C, Mazzei D, et al. (2016) Non-Ergot-Dopamine Agonists don't Increase the Risk of Heart Failure in Parkinson's disease Patients: A Meta-Analysis of Randomized Controlled Trials. J Clin Med Res 8: 449-460. doi: 10.14740/jocmr2541e
|
| [25] |
Evangelista I, Nuti R, Picchioni T, et al. (2019) Molecular Dysfunction and Phenotypic Derangement in Diabetic Cardiomyopathy. Int J Mol Sci 20: 3264. doi: 10.3390/ijms20133264
|
| [26] | Tank AW, Lee WD (2015) Peripheral and central effects of circulating catecholamine's. Compr Physiol 5: 1-15. |
| [27] | Duarte AI, Moreira PI, Oliveira CR (2012) Insulin in central nervous system: more than just a peripheral hormone. J Aging Res 384: 1414-1431. |
| [28] | Zheng J, Wang Y, Han S, et al. (2018) Identification of Protein Kinase C Isoforms Involved in Type 1 Diabetic Encephalopathy in Mice. J Diabetes Res 8431249. |
| [29] |
Das SK, Yuan YF, Li MQ (2018) Specific PKC βII inhibitor: one stone two birds in the treatment of diabetic foot ulcers. Biosci Rep 38: BSR20171459. doi: 10.1042/BSR20171459
|
| [30] | Nokkaew N, Mongkolpathumrat P, Junsiri R, et al. (2019) p38 MAPK Inhibitor (SB203580) and Metformin Reduces Aortic Protein Carbonyl and Inflammation in Non-obese Type 2 Diabetic Rats. Ind J Clin Biochem 1–7. |
| [31] | Nokkaew N, Sanit J, Mongkolpathumrat P, et al. (2019) Anti-diabetic drug, metformin, and the p38 inhibitor (SB203580) reduces internal organs oxidative stress in non-obese type 2 diabetic rats. J Appl Pharm Sci 9: 12-20. |
| [32] |
Cramer SC, Sur M, Dobkin BH, et al. (2011) Harnessing neuroplasticity for clinical applications. Brain 134: 1591-1609. doi: 10.1093/brain/awr039
|
| [33] |
Hui C, Jingli L, Jiao D, et al. (2015) TAK1 inhibition prevents the development of autoimmune diabetes in NOD mice. Sci Rep 5: 14593. doi: 10.1038/srep14593
|
| [34] |
Luchsinger JA, Reitz C, Patel B, et al. (2007) Mayeux, Relation of diabetes to mild cognitive impairment. Arch Neurol 64: 570-575. doi: 10.1001/archneur.64.4.570
|
| [35] |
Carvalho C, Cardoso S, Correia SC, et al. (2012) Metabolic alterations induced by sucrose intake and Alzheimer's disease promote similar brain mitochondrial abnormalities. Diabetes 61: 1234-1242. doi: 10.2337/db11-1186
|
| [36] |
Bell DS (2003) Diabetic cardiomyopathy. Diabetes Care 26: 2949-2951. doi: 10.2337/diacare.26.10.2949
|
| [37] |
Tschope C, Walther T, Koniger J, et al. (2004) Prevention of cardiac fibrosis and left ventricular dysfunction in diabetic cardiomyopathy in rats by transgenic expression of the human tissue kallikrein gene. Faseb J 18: 828-835. doi: 10.1096/fj.03-0736com
|
| [38] |
Fischer TA, Ludwig S, Flory E, et al. (2001) Activation of cardiac c-Jun NH(2)-terminal kinases and p38-mitogen-activated protein kinases with abrupt changes in hemodynamic load. Hypertension 37: 1222-1228. doi: 10.1161/01.HYP.37.5.1222
|
| [39] |
Zhang GX, Kimura S, Nishiyama A, et al. (2004) ROS during the acute phase of Ang-II hypertension participates in cardiovascular MAPK activation but not vasoconstriction. Hypertension 43: 117-124. doi: 10.1161/01.HYP.0000105110.12667.F8
|
| [40] |
Steendijk P, Staal E, Jukema JW, et al. (2001) Hypertonic saline method accurately determines parallel conductance for dual-field conductance catheter. Am J Physiol Heart Circ Physiol 281: H755-H763. doi: 10.1152/ajpheart.2001.281.2.H755
|
| [41] |
Steenbergen C (2002) The role of p38 mitogen-activated protein kinase in myocardial ischemia/reperfusion injury; relationship to ischemic preconditioning. Basic Res Cardiol 97: 276-285. doi: 10.1007/s00395-002-0364-9
|
| [42] |
Gorog DA, Tanno M, Cao X, et al. (2004) Inhibition of p38 MAPK activity fails to attenuate contractile dysfunction in a mouse model of low-flow ischemia. Cardiovasc Res 61: 123-131. doi: 10.1016/j.cardiores.2003.09.034
|
| [43] |
Westermann D, Rutschow S, Van Linthout S, et al. (2006) Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia 49: 2507-2513. doi: 10.1007/s00125-006-0385-2
|
| [44] |
Pereira S, Yu WQ, Moore J, et al. (2016) Effect of a p38 MAPK inhibitor on FFA-induced hepatic insulin resistance in-vivo. Nutr Diabetes 6: e210. doi: 10.1038/nutd.2016.11
|
| [45] | Xu J, Li J, Hou R, et al. (2019) JPQ downregulates the P38 MAPK signal pathway in skeletal muscle of diabetic rats and increases the insulin sensitivity of Skeletal Muscle. Int J Clin Exp Med 12: 5130-5137. |
| [46] |
Erik V, Marjut L, Hanna F, et al. (2010) Sirtuin1-p53, forkhead box O3a, p38 and post-infarct cardiac remodeling in the spontaneously diabetic Goto-Kakizaki rat. Cardiovasc Diabetol 9: 1-13. doi: 10.1186/1475-2840-9-1
|
| [47] |
Wang S, Ding L, Ji H, et al. (2016) The Role of p38 MAPK in the Development of Diabetic Cardiomyopathy. Int J Mol Sci 17: 1037. doi: 10.3390/ijms17071037
|
| [48] | Jantira S, Eakkapote P, Punyanuch A, et al. (2019) Combination of metformin and p38 MAPK inhibitor, SB203580, reduced myocardial ischemia/reperfusion injury in non-obese type 2 diabetic Goto-Kakizaki rats. Exp Ther Med 18: 1701-1714. |
| [49] |
Xie D, Zhao J, Guo R (2020) Sevoflurane pre-conditioning ameliorates diabetic myocardial ischemia/reperfusion injury via differential regulation of p38 and ERK. Sci Rep 10: 23. doi: 10.1038/s41598-019-56897-8
|
| [50] |
Gao F, Yue TL, Shi DW, et al. (2002) p38 MAPK inhibition reduces myocardial reperfusion injury via inhibition of endothelial adhesion molecule expression and blockade of PMN accumulation. Cardiovasc Res 53: 414-422. doi: 10.1016/S0008-6363(01)00488-6
|
| [51] |
Dubash AD, Kam CY, Aguado BA, et al. (2016) Plakophilin-2 loss promotes TGF-β1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes. J Cell Biol 212: 425. doi: 10.1083/jcb.201507018
|
| [52] |
Umbarkar P, Singh AP, Gupte M, et al. (2019) Cardiomyocyte SMAD4-Dependent TGF-β Signaling is Essential to Maintain Adult Heart Homeostasis. JACC Basic Transl Sci 4: 41-53. doi: 10.1016/j.jacbts.2018.10.003
|
| [53] |
Palojoki E, Saraste A, Eriksson A, et al. (2001) Cardiomyocyte apoptosis and ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol 280: H2726-H2731. doi: 10.1152/ajpheart.2001.280.6.H2726
|
| [54] | Dong H, Cui B, Hao X (2019) MicroRNA-22 alleviates inflammation in ischemic stroke via p38 MAPK pathways. Mol Med Rep 20: 735-744. |
| [55] | Yu L, Li Z, Dong X, et al. (2018) Polydatin Protects Diabetic Heart against Ischemia-Reperfusion Injury via Notch1/Hes1-Mediated Activation of Pten/Akt Signaling. Oxid Med Cell Longev 2018: 2750695. |
| [56] |
Stockand JD, Meszaros JG (2003) Aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-Ras A and MAPK1/2 signaling. Am J Physiol Heart Circ Physiol 284: H176-H184. doi: 10.1152/ajpheart.00421.2002
|
| [57] |
Koga Y, Tsurumaki H, Aoki-Saito H, et al. (2019) Roles of Cyclic AMP Response Element Binding Activation in the ERK1/2 and p38 MAPK Signaling Pathway in Central Nervous System, Cardiovascular System, Osteoclast Differentiation and Mucin and Cytokine Production. Int J Mol Sci 20: 1346. doi: 10.3390/ijms20061346
|
| [58] |
Turner NA, Blythe NM (2019) Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling. J Cardiovasc Dev Dis 6: 27. doi: 10.3390/jcdd6030027
|
| [59] |
Thum T, Gross C, Fiedler J, et al. (2008) Micro RNA-21 contributes to myocardial disease by stimulating MAP kinase signaling in fibroblasts. Nature 456: 980-984. doi: 10.1038/nature07511
|
| [60] |
Liang Q, Molkentin JD (2003) Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol 35: 1385-1394. doi: 10.1016/j.yjmcc.2003.10.001
|
| [61] |
Xu Z, Sun J, Tong Q, et al. (2016) The Role of ERK1/2 in the Development of Diabetic Cardiomyopathy. Int J Mol Sci 17: 2001. doi: 10.3390/ijms17122001
|
| [62] |
Ruiz M, Coderre L, Allen BG, et al. (2018) Protecting the heart through MK2 modulation, toward a role in diabetic cardiomyopathy and lipid metabolism. Biochim Biophys Acta Mol Basis Dis 1864: 1914-1922. doi: 10.1016/j.bbadis.2017.07.015
|
| [63] |
Liao P, Georgakopoulos D, Kovacs A, et al. (2001) The in-vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A 98: 12283-12288. doi: 10.1073/pnas.211086598
|
| [64] |
Jia G, Hill MA, Sowers JR (2018) Diabetic Cardiomyopathy An Update of Mechanisms Contributing to This Clinical Entity. Circ Res 122: 624-638. doi: 10.1161/CIRCRESAHA.117.311586
|
| [65] |
Streicher JM, Ren S, Herschman H, et al. (2010) MAPK-activated protein kinase-2 in cardiac hypertrophy and cyclooxygenase-2 regulation in heart. Circ Res 106: 1434-1443. doi: 10.1161/CIRCRESAHA.109.213199
|
| [66] |
Hill JA, Olson EN (2008) Cardiac plasticity. N Engl J Med 358: 1370-1380. doi: 10.1056/NEJMra072139
|
| [67] |
Vikas K, Kumar A, Rahul S, et al. (2019) Chronic Pressure Overload Results in Deficiency of Mitochondrial Membrane Transporter ABCB7 Which Contributes to Iron Overload, Mitochondrial Dysfunction, Metabolic Shift and Worsens Cardiac Function. Sci Rep 9: 13170. doi: 10.1038/s41598-019-49666-0
|
| [68] |
Takeda N, Manabe I, Uchino Y, et al. (2010) Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest 120: 254-265. doi: 10.1172/JCI40295
|
| [69] |
Small EM (2012) The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J Cardiovasc Transl Res 5: 794-804. doi: 10.1007/s12265-012-9397-0
|
| [70] |
Zent J, Guo LW (2018) Signaling Mechanisms of Myofibroblastic Activation: Outside-in and Inside-Out. Cell Physiol Biochem 49: 848-868. doi: 10.1159/000493217
|
| [71] |
Bujak M, Frangogiannis NG (2007) The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 74: 184-195. doi: 10.1016/j.cardiores.2006.10.002
|
| [72] |
Ieda M, Tsuchihashi T, Ivey KN, et al. (2009) Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev Cell 16: 233-244. doi: 10.1016/j.devcel.2008.12.007
|
| [73] |
Souders CA, Bowers SL, Baudino TA (2009) Cardiac fibroblast: the renaissance cell. Circ Res 105: 1164-1176. doi: 10.1161/CIRCRESAHA.109.209809
|
| [74] |
Kakkar R, Lee RT (2010) Intramyocardial fibroblast myocyte communication. Circ Res 106: 47-57. doi: 10.1161/CIRCRESAHA.109.207456
|
| [75] |
Martin ML, Blaxall BC (2012) Cardiac intercellular communication: are myocytes and fibroblasts fair-weather friends? J Cardiovasc Transl Res 5: 768-782. doi: 10.1007/s12265-012-9404-5
|
| [76] |
Furtado MB, Costa MW, Pranoto EA, et al. (2014) Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ Res 114: 1422-1434. doi: 10.1161/CIRCRESAHA.114.302530
|
| [77] |
Miyazaki T, Haraguchi S, Kim-Kaneyama JR, et al. (2019) Endothelial calpain systems orchestrate myofibroblast differentiation during wound healing. FASEB J 33: fj.201800588RR. doi: 10.1096/fj.201800588RR
|
| [78] |
Zhang ZY, Wang N, Qian LL, et al. (2020) Glucose fluctuations promote aortic fibrosis through the ROS/p38 MAPK/Runx2 signaling pathway. J Vasc Res 57: 24-33. doi: 10.1159/000503608
|
| [79] |
Nian M, Lee P, Khaper N, et al. (2004) Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res 94: 1543-1553. doi: 10.1161/01.RES.0000130526.20854.fa
|
| [80] | Lee MMY, McMurray JJV, Lorenzo-Almorós A, et al. (2016) Diabetic cardiomyopathy. Heart 105. |
| [81] |
Kocabaş U, Yılmaz Ö, Kurtoğlu V (2019) Diabetic cardiomyopathy: acute and reversible left ventricular systolic dysfunction due to cardiotoxicity of hyperglycaemic hyperosmolar state—a case report. Eur Heart J Case Rep 3: ytz049. doi: 10.1093/ehjcr/ytz049
|
| [82] |
Gao M, Wang X, Zhang X, et al. (2015) Attenuation of Cardiac Dysfunction in Polymicrobial Sepsis by MicroRNA-146a Is Mediated via Targeting of IRAK1 and TRAF6 Expression. J Immunol 195: 672-682. doi: 10.4049/jimmunol.1403155
|
| [83] |
Mann DL (2003) Stress-activated cytokines and the heart: from adaptation to maladaptation. Annu Rev Physiol 65: 81-101. doi: 10.1146/annurev.physiol.65.092101.142249
|
| [84] |
Fiordelisi A, Iaccarino G, Morisco C, et al. (2019) NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int J Mol Sci 20: 1599. doi: 10.3390/ijms20071599
|
| [85] |
Frati G, Schirone L, Chimenti I, et al. (2017) An overview of the inflammatory signaling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc Res 113: 378-388. doi: 10.1093/cvr/cvx011
|
| [86] |
Sharov VG, Todor A, Suzuki G, et al. (2003) Hypoxia, angiotensin-II, and norepinephrine mediated apoptosis is stimulus specific in canine failed cardiomyocytes: a role for p38 MAPK, Fas-L and cyclin D1. Eur J Heart Fail 5: 121-129. doi: 10.1016/S1388-9842(02)00254-4
|
| [87] |
Kaiser RA, Bueno OF, Lips DJ, et al. (2004) Targeted inhibition of p38 mitogen-activated protein kinase antagonizes cardiac injury and cell death following ischemia-reperfusion in-vivo. J Biol Chem 279: 15524-15530. doi: 10.1074/jbc.M313717200
|
| [88] |
Wakeman D, Guo J, Santos JA, et al. (2012) p38 MAPK regulates Bax activity and apoptosis in enterocytes at baseline and after intestinal resection. Am J Physiol Gastrointest Liver Physiol 302: G997-1005. doi: 10.1152/ajpgi.00485.2011
|
| [89] |
Xu Q, Fang H, Zhao L, et al. (2019) Mechano growth factor attenuates mechanical overload-induced nucleus pulposus cell apoptosis through inhibiting the p38 MAPK pathway. Biosci Rep 39: BSR20182462. doi: 10.1042/BSR20182462
|
| [90] |
Aggeli IK, Beis I, Gaitanaki C (2008) Oxidative stress and calpain inhibition induces alpha B-crystallin phosphorylation via p38 MAPK and calcium signaling pathways in H9c2 cells. Cell Signal 20: 1292-1302. doi: 10.1016/j.cellsig.2008.02.019
|
| [91] |
Mitra A, Ray A, Datta R, et al. (2014) Cardioprotective role of P38 MAPK during myocardial infarction via parallel activation of alpha-crystallin B and Nrf2. J Cell Physiol 229: 1272-1282. doi: 10.1002/jcp.24565
|
| [92] |
Kim JK, Pedram A, Razandi M, et al. (2006) Estrogen prevents cardiomyocyte apoptosis through inhibition of reactive oxygen species and differential regulation of p38 kinase isoforms. J Biol Chem 281: 6760-6767. doi: 10.1074/jbc.M511024200
|
| [93] |
Liu H, Pedram A, Kim JK (2011) Oestrogen prevents cardiomyocyte apoptosis by suppressing p38alpha-mediated activation of p53 and by down-regulating p53 inhibition on p38beta. Cardiovasc Res 89: 119-128. doi: 10.1093/cvr/cvq265
|
| [94] |
Wu H, Wang G, Li S, et al. (2015) TNF-α- Mediated-p38-Dependent Signaling Pathway Contributes to Myocyte Apoptosis in Rats Subjected to Surgical Trauma. Cell Physiol Biochem 35: 1454-1466. doi: 10.1159/000373965
|
| [95] |
Zuo G, Ren X, Qian X, et al. (2019) Inhibition of JNK and p38 MAPK-mediated inflammation and apoptosis by ivabradine improves cardiac function in streptozotocin-induced diabetic cardiomyopathy. J Cell Physiol 234: 1925-1936. doi: 10.1002/jcp.27070
|
| [96] |
Li Z, Ma JY, Kerr I, et al. (2006) Selective inhibition of p38alpha MAPK improves cardiac function and reduces myocardial apoptosis in rat model of myocardial injury. Am J Physiol Heart Circ Physiol 291: H1972-H1977. doi: 10.1152/ajpheart.00043.2006
|
| [97] |
Adhikary L, Chow F, Nikolic-Paterson DJ, et al. (2004) Abnormal p38 mitogen-activated protein kinase signaling in human and experimental diabetic nephropathy. Diabetologia 47: 1210-1222. doi: 10.1007/s00125-004-1437-0
|
| [98] |
Kojonazarov B, Novoyatleva T, Boehm M, et al. (2017) p38 MAPK Inhibition Improves Heart Function in Pressure-Loaded Right Ventricular Hypertrophy. Am J Respir Cell Mol Biol 57: 603-614. doi: 10.1165/rcmb.2016-0374OC
|
| [99] |
Seeger FH, Sedding D, Langheinrich AC, et al. (2010) Inhibition of the p38 MAP kinase in-vivo improves number and functional activity of vasculogenic cells and reduces atherosclerotic disease progression. Basic Res Cardiol 105: 389-397. doi: 10.1007/s00395-009-0072-9
|
| [100] |
Nediani C, Borchi E, Giordano C, et al. (2007) NADPH oxidase-dependent redox signaling in human heart failure: relationship between the left and right ventricle. J Mol Cell Cardiol 42: 826-834. doi: 10.1016/j.yjmcc.2007.01.009
|
| [101] |
Newby LK, Marber MS, Melloni C, et al. (2014) SOLSTICE Investigators. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: a randomised phase 2 trial. Lancet 384: 1187-1195. doi: 10.1016/S0140-6736(14)60417-7
|
| [102] |
Halpern CH, Tekriwal A, Santollo J, et al. (2013) Amelioration of binge eating by nucleus accumbens shell deep brain stimulation in mice involves D2 receptor modulation. J Neurosci 33: 7122-7129. doi: 10.1523/JNEUROSCI.3237-12.2013
|
| [103] |
Ter Horst KW, Lammers NM, Trinko R, et al. (2018) Striatal dopamine regulates systemic glucose metabolism in humans and mice. Sci Transl Med 10: eaar3752. doi: 10.1126/scitranslmed.aar3752
|
| [104] |
Figee M, De Koning P, Klaassen S, et al. (2014) Deep brain stimulation induces striatal dopamine release in obsessive-compulsive disorder. Biol Psychiatry 75: 647-652. doi: 10.1016/j.biopsych.2013.06.021
|
| [105] |
Boot E, Booij J, Hasler G, et al. (2008) AMPT-induced monoamine depletion in humans: Evaluation of two alternative (123I) IBZM SPECT procedures. Eur J Nucl Med Mol Imaging 35: 1350-1356. doi: 10.1007/s00259-008-0739-8
|
| [106] |
Zeng C, Zhang M, Asico LD, et al. (2007) The dopaminergic system in hypertension. Clin Sci 112: 583-597. doi: 10.1042/CS20070018
|
| [107] |
Channabasappa S, Sanjay K (2011) Bromocriptine in type 2 diabetes mellitus. Indian J Endocrinol Metab 15: S17-S24. doi: 10.4103/2230-8210.83058
|
| [108] |
Reda E, Hassaneen S, El-Abhar HS (2018) Novel Trajectories of Bromocriptine Antidiabetic Action: Leptin-IL-6/ JAK2/p-STAT3/SOCS3, p-IR/p-AKT/GLUT4, PPAR-γ/Adiponectin, Nrf2/PARP-1, and GLP-1. Front Pharmacol 9: 771. doi: 10.3389/fphar.2018.00771
|
| [109] |
Leicht M, Briest W, Zimmer HG (2003) Regulation of norepinephrine-induced proliferation in cardiac fibroblasts by interleukin-6 and p42/p44 mitogen activated protein kinase. Mol Cell Biochem 243: 65-72. doi: 10.1023/A:1021655023870
|
| [110] |
Lubahn CL, Lorton D, Schaller JA, et al. (2014) Targeting a-and b-adrenergic receptors differentially shift Th1, Th2, and inflammatory cytokine profiles in immune organs to attenuate adjuvant arthritis. Front Immunol 5: 346. doi: 10.3389/fimmu.2014.00346
|
| [111] |
Moliner P, Enjuanes C, Tajes M, et al. (2019) Association Between Norepinephrine Levels and Abnormal Iron Status in Patients With Chronic Heart Failure: Is Iron Deficiency More Than a Comorbidity? J Am Heart Assoc 8: e010887. doi: 10.1161/JAHA.118.010887
|
| [112] |
Zhang P, Li Y, Nie K, et al. (2018) Hypotension and bradycardia, a serious adverse effect of piribedil, a case report and literature review. BMC Neurol 18: 221. doi: 10.1186/s12883-018-1230-1
|
| [113] | Michael E, Shuqin L, Nicholas C, et al. (2019) 1793-P: Dopamine D1 plus D2 Receptor Coactivation Ameliorates Metabolic Syndrome (MS) and Nonalcoholic Fatty Liver Disease (NAFLD) in Mice. Diabetes 68. |
| [114] |
Monti JM, Monti D (2007) The involvement of dopamine in the modulation of sleep and waking. Sleep Med Rev 11: 113-133. doi: 10.1016/j.smrv.2006.08.003
|
| [115] |
Wang X, Wang ZB, Luo C, et al. (2019) The Prospective Value of Dopamine Receptors on Bio-Behavior of Tumor. J Cancer 10: 1622-1632. doi: 10.7150/jca.27780
|
| [116] |
Chávez-Castillo M, Ortega Á, Nava M, et al. (2018) Metabolic risk in depression and treatment with selective serotonin reuptake inhibitors: are the metabolic syndrome and an increase in cardiovascular risk unavoidable? Vessel Plus 2: 6. doi: 10.20517/2574-1209.2018.02
|
| [117] | Fortier JH, Pizzarotti B, Shaw RE, et al. (2019) Drug-associated valvular heart diseases and serotonin-related pathways: a meta-analysis. Heart 105: 1140-1148. |
| [118] |
Mawe GM, Hoffman JM (2013) Serotonin signaling in the gastrointestinal tract. Nat Rev Gastroenterol Hepatol 10: 473-486. doi: 10.1038/nrgastro.2013.105
|
| [119] |
Selim AM, Sarswat N, Kelesidis I, et al. (2017) Plasma Serotonin in Heart Failure: Possible Marker and Potential Treatment Target. Heart Lung Circ 26: 442-449. doi: 10.1016/j.hlc.2016.08.003
|
| [120] |
Guo S, Chen L, Cheng S, et al. (2019) Comparative cardiovascular safety of selective serotonin reuptake inhibitors (SSRIs) among Chinese senile depression patients: A network meta-analysis of randomized controlled trials. Medicine 98: e15786. doi: 10.1097/MD.0000000000015786
|
| [121] |
Lancellotti P, Nchimi A, Hego A, et al. (2015) High-dose oral intake of serotonin induces valvular heart disease in rabbits. Int J Cardiol 197: 72-75. doi: 10.1016/j.ijcard.2015.06.035
|
| [122] |
Seferovic PM, Ponikowski P, Anker SD, et al. (2019) Clinical practice update on heart failure 2019: pharmacotherapy, procedures, devices and patient management. An expert consensus meeting report of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 21: 1169-1186. doi: 10.1002/ejhf.1531
|
| [123] |
Shimizu Y, Minatoguchi S, Hashimoto K, et al. (2002) The role of serotonin in ischemic cellular damage and the infarct size-reducing effect of sarpogrelate, a 5-hydroxytryptamine-2 receptor blocker, in rabbit hearts. J Am Coll Cardiol 40: 1347-1355. doi: 10.1016/S0735-1097(02)02158-7
|
| [124] |
Chen YG, Mathews CE, Driver JP (2018) The Role of NOD Mice in Type 1 Diabetes Research: Lessons from the Past and Recommendations for the Future. Front Endocrinol 9: 51. doi: 10.3389/fendo.2018.00051
|
| [125] |
Pei Y, Cui F, Du X, et al. (2019) Antioxidative nanofullerol inhibits macrophage activation and development of osteoarthritis in rats. Int J Nanomedicine 14: 4145-4155. doi: 10.2147/IJN.S202466
|
| [126] |
Kullmann S, Heni M, Hallschmid M, et al. (2016) Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol Rev 96: 169-209. doi: 10.1152/physrev.00032.2015
|
| [127] |
Grillo CA, Woodruff JL, Macht VA, et al. (2019) Insulin resistance and hippocampal dysfunction: Disentangling peripheral and brain causes from consequences. Exp Neurol 318: 71-77. doi: 10.1016/j.expneurol.2019.04.012
|
| [128] |
Nakabeppu Y (2019) Origins of brain insulin and its function. In: Diabetes Mellitus. Advances in Experimental Medicine and Biology. Adv Exp Med Biol 1128: 1-11. doi: 10.1007/978-981-13-3540-2_1
|
| [129] |
Bode BW, Garg SK (2016) The Emerging Role of Adjunctive Noninsulin Anti-hyperglycemic Therapy in the Management of Type 1 Diabetes. Endocr Pract 22: 220-230. doi: 10.4158/EP15869.RA
|
| [130] | Otto-Buczkowska E, Nowowiejska B, Jarosz-Chobot P, et al. (2009) Could oral antidiabetic agents be useful in the management of different types of diabetes and syndromes of insulin resistance in children and adolescents? Przegl Lek 66: 388-393. |
| [131] | Otto-Buczkowska E, Natalia J (2018) Pharmacological Treatment in Diabetes Mellitus Type 1 – Insulin and What Else? Int J Endocrinol Metab 16: e13008. |
| [132] |
Grizzanti J, Corrigan R, Casadesusa G (2018) Neuroprotective Effects of Amylin Analogues on Alzheimer's Disease Pathogenesis and Cognition. J Alzheimers Dis 66: 11-23. doi: 10.3233/JAD-180433
|
| [133] |
Alicic RZ, Neumiller JJ, Johnson EJ, et al. (2019) Sodium-Glucose Cotransporter 2 Inhibition and Diabetic Kidney Disease. Diabetes 68: 248-257. doi: 10.2337/dbi18-0007
|
| [134] |
Mullane K, Williams M (2019) Preclinical Models of Alzheimer's Disease: Relevance and Translational Validity. Curr Protoc Pharmacol 84: e57. doi: 10.1002/cpph.57
|
| [135] |
Antal Z, Baker JC, Smith C, et al. (2012) Beyond HLA-A*0201: new HLA-transgenic non-obese diabetic mouse models of type 1 diabetes identify the insulin C-peptide as a rich source of CD8+T cell epitopes. J Immunol 188: 5766-5775. doi: 10.4049/jimmunol.1102930
|
| [136] |
Serr P, Santamaria P (2019) Antigen-specific therapeutic approaches for autoimmunity. Nature Biotechnol 37: 238-251. doi: 10.1038/s41587-019-0015-4
|
| [137] |
Singer-Englar T, Barlow G, Mathur R (2018) Obesity, diabetes, and the gut microbiome: an updated review. Expert Rev Gastroenterol Hepatol 13: 3-15. doi: 10.1080/17474124.2019.1543023
|
| [138] |
Siljandera H, Honkanenb J, Knipa M (2019) Microbiome and type 1 diabetes. Ebiomedicine 46: 512-521. doi: 10.1016/j.ebiom.2019.06.031
|
| [139] |
Escós A, Risco A, Alsina-Beauchamp D, et al. (2016) p38γ and p38δ Mitogen Activated Protein Kinases (MAPKs), New Stars in the MAPK Galaxy. Front Cell Dev Biol 4: 31. doi: 10.3389/fcell.2016.00031
|
| [140] |
Cuenda A, Rousseau S (2007) p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta 1773: 1358-1375. doi: 10.1016/j.bbamcr.2007.03.010
|
| [141] |
Beardmore VA, Hinton HJ, Eftychi C, et al. (2005) Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol Cell Biol 25: 10454-10464. doi: 10.1128/MCB.25.23.10454-10464.2005
|
| [142] |
Remy G, Risco AM, Iñesta-Vaquera FA, et al. (2010) Differential activation of p38 MAPK isoforms by MKK6 and MKK3. Cell Signal 22: 660-667. doi: 10.1016/j.cellsig.2009.11.020
|
| [143] |
Jiang Y, Gram H, Zhao M, et al. (1997) Characterization of the structure and function of the fourth member of p38 group mitogen activated protein kinases, p38δ. J Biol Chem 272: 30122-30128. doi: 10.1074/jbc.272.48.30122
|
| [144] |
Sumara G, Formentini I, Collinsetal S (2009) Regulation of PKD by the MAPK p38 delta in insulin secretion and glucose homeostasis. Cell 136: 235-248. doi: 10.1016/j.cell.2008.11.018
|
| [145] |
Lee JC, Laydon JT, McDonnelletal PC (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372: 739-746. doi: 10.1038/372739a0
|
| [146] |
Jiang Y, Chen C, Li Z, et al. (1996) Characterization of the structure and function of a new mitogen activated protein kinase (p38β). J Biol Chem 271: 17920-17926. doi: 10.1074/jbc.271.30.17920
|
| [147] |
Cuenda A, Rouse J, Dozaetal YN (1995) SB203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Letters 364: 229-233. doi: 10.1016/0014-5793(95)00357-F
|
| [148] |
Ramachandra CJ, Mehta A, Wong P, et al. (2016) ErbB4 Activated p38gamma MAPK isoform mediates early cardiogenesis through NKx2.5 in human pluripotent stem cells. Stem Cells 34: 288-298. doi: 10.1002/stem.2223
|
| [149] |
González-Terán B, López JA, Rodríguez E, et al. (2016) p38gamma and delta promote heart hypertrophy by targeting the mTOR-inhibitory protein DEPTOR for degradation. Nat Commun 7: 10477. doi: 10.1038/ncomms10477
|
| [150] |
Cuevas BD, Abell AN, Johnson GL (2007) Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene 26: 3159-3171. doi: 10.1038/sj.onc.1210409
|
| [151] |
Cuenda A, Rousseau S (2007) p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta 1773: 1358-1375. doi: 10.1016/j.bbamcr.2007.03.010
|
| [152] |
Chang CI, Xu BE, Akella R, et al. (2002) Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol Cell 9: 1241-1249. doi: 10.1016/S1097-2765(02)00525-7
|
| [153] |
Biondi RM, Nebreda AR (2003) Signaling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem J 372: 1-13. doi: 10.1042/bj20021641
|
| [154] |
Enslen H, Brancho DM, Davis RJ (2000) Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J 19: 1301-1311. doi: 10.1093/emboj/19.6.1301
|
| [155] |
Tomlinson DR (1999) Mitogen-activated protein kinases as glucose transducers for diabetic complications. Diabetologia 42: 1271-1281. doi: 10.1007/s001250051439
|
| [156] |
Begum N, Ragolia L (2000) High glucose and insulin inhibit VSMC MKP-1 expression by blocking iNOS via p38 MAPK activation. Am J Physiol Cell Physiol 278: C81-C91. doi: 10.1152/ajpcell.2000.278.1.C81
|
| [157] |
Chen S, Qiong Y, Gardner DG (2006) Aroleforp38mitogen-activatedproteinkinase and c-Myc inendothelin-dependent rat aortic smooth muscle cell proliferation. Hypertension 47: 252-258. doi: 10.1161/01.HYP.0000198424.93598.6b
|
| [158] |
Natarajan R, Scott S, Bai W, et al. (1999) Angiotensin II signaling in vascular smoothmuscle cells under high glucose conditions. Hypertension 33: 378-384. doi: 10.1161/01.HYP.33.1.378
|
| [159] |
Igarashi M, Wakasaki H, Takahara N, et al. (1999) Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways. J Clin Invest 103: 185-195. doi: 10.1172/JCI3326
|
| [160] |
Dorenkamp M, Riad AS, Stiehl S, et al. (2005) Protection against oxidative stress indiabetic rats: role of angiotensinAT1 receptor and beta 1-adrenoceptor antagonism. Eur J Pharmacol 520: 179-187. doi: 10.1016/j.ejphar.2005.07.020
|
| [161] |
Begum N, Ragolia L (2000) High glucose and insulin inhibit VSMC MKP-1 expression by blocking iNOS via p38 MAPK activation. Am J Physiol Cell Physiol 278: C81-C91. doi: 10.1152/ajpcell.2000.278.1.C81
|
| [162] |
Chen S, Qiong Y, Gardner DG (2006) A role for p38 mitogen-activated protein kinase and c-Myc in endothelin-dependent rat aortic smooth muscle cell proliferation. Hypertension 47: 252-258. doi: 10.1161/01.HYP.0000198424.93598.6b
|
| [163] |
Cain BS, Meldrum DR, Meng X, et al. (1999) p38 MAPK inhibition decreases TNF-α production and enhances post ischemic human myocardial function. J Surg Res 83: 7-12. doi: 10.1006/jsre.1998.5548
|
| [164] |
Communal C, Colucci WS, Singh K (2000) p38 mitogen-activated protein kinase pathway protects adult rat ventricular myocytes againstβ-adrenergic receptor-stimulated apoptosis. Evidence for Gi-dependent activation. J Biol Chem 275: 19395-19400. doi: 10.1074/jbc.M910471199
|
| [165] |
Liang Q, Molkentin JD (2003) Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol 35: 1385-1394. doi: 10.1016/j.yjmcc.2003.10.001
|
| [166] |
Li M, Georgakopoulos D, Luetal G (2005) p38MAPkinase mediates inflammatory cytokine induction in cardiomyocytes and extracellular matrix remodeling in heart. Circulation 111: 2494-2502. doi: 10.1161/01.CIR.0000165117.71483.0C
|
| [167] | Hu SS, Kong LZ, Gaoetal RL (2010) Outline of the report on cardiovascular disease in China. Biomed Environ Sci 25: 251-256. |
| [168] |
Yang HS, Zheng QY, Duetal YY (2016) Influence of different acupoint combinations on immediate effect of surface electromyography of patients with cervical spondylosis. World J Acupunct Moxibustion 26: 7-13. doi: 10.1016/S1003-5257(17)30056-9
|
| [169] |
Pan YX, Chen KF, Lin YX, et al. (2013) Intracisternal administration of SB203580, a p38 mitogen-activated protein kinase tumor necrosis factor-alpha. J Clin Neurosci 20: 726-730. doi: 10.1016/j.jocn.2012.09.012
|
| [170] | Wu S, Li J, Hong YQ, et al. (2012) Efects of electroacupuncture at Neiguan (PC 6) on p38 MAPK signaling pathway in rats with cardiac hypertrophy. Chin Acupunct Moxibustion 32: 145-148. |
| [171] |
Du Y, Tang J, Li G, et al. (2010) Effects of p38 MAPK Inhibition on Early Stages of Diabetic Retinopathy and Sensory Nerve Function. Invest Ophthalmol Vis Sci 51: 2158-2164. doi: 10.1167/iovs.09-3674
|
| [172] |
Wang S, Ding L, Zheng Y, et al. (2016) The role of p38 MAPK in the development of diabetic cardiomyopathy. Int J Mol Sci 17: 1037. doi: 10.3390/ijms17071037
|
| [173] |
Muslin AJ (2008) MAPK signaling in cardiovascular health and disease: Molecular mechanisms and therapeutic targets. Clin Sci (Lond) 115: 203-218. doi: 10.1042/CS20070430
|
| [174] |
Radi ZA, Marusak RA, Morris DL (2009) Species comparison of the role of p38 MAPK in the female reproductive system. J Toxicol Pathol 22: 109-124. doi: 10.1293/tox.22.109
|
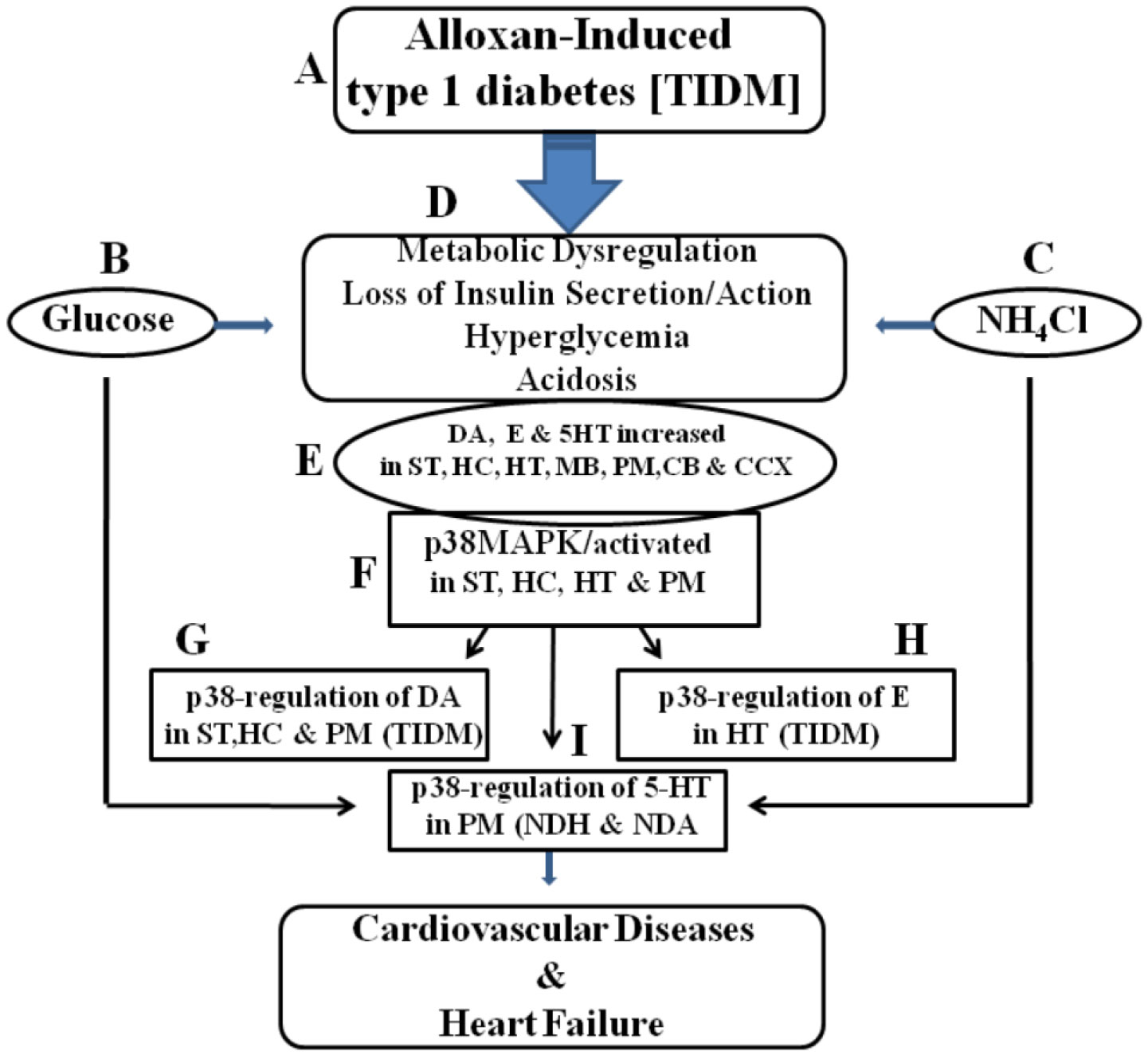
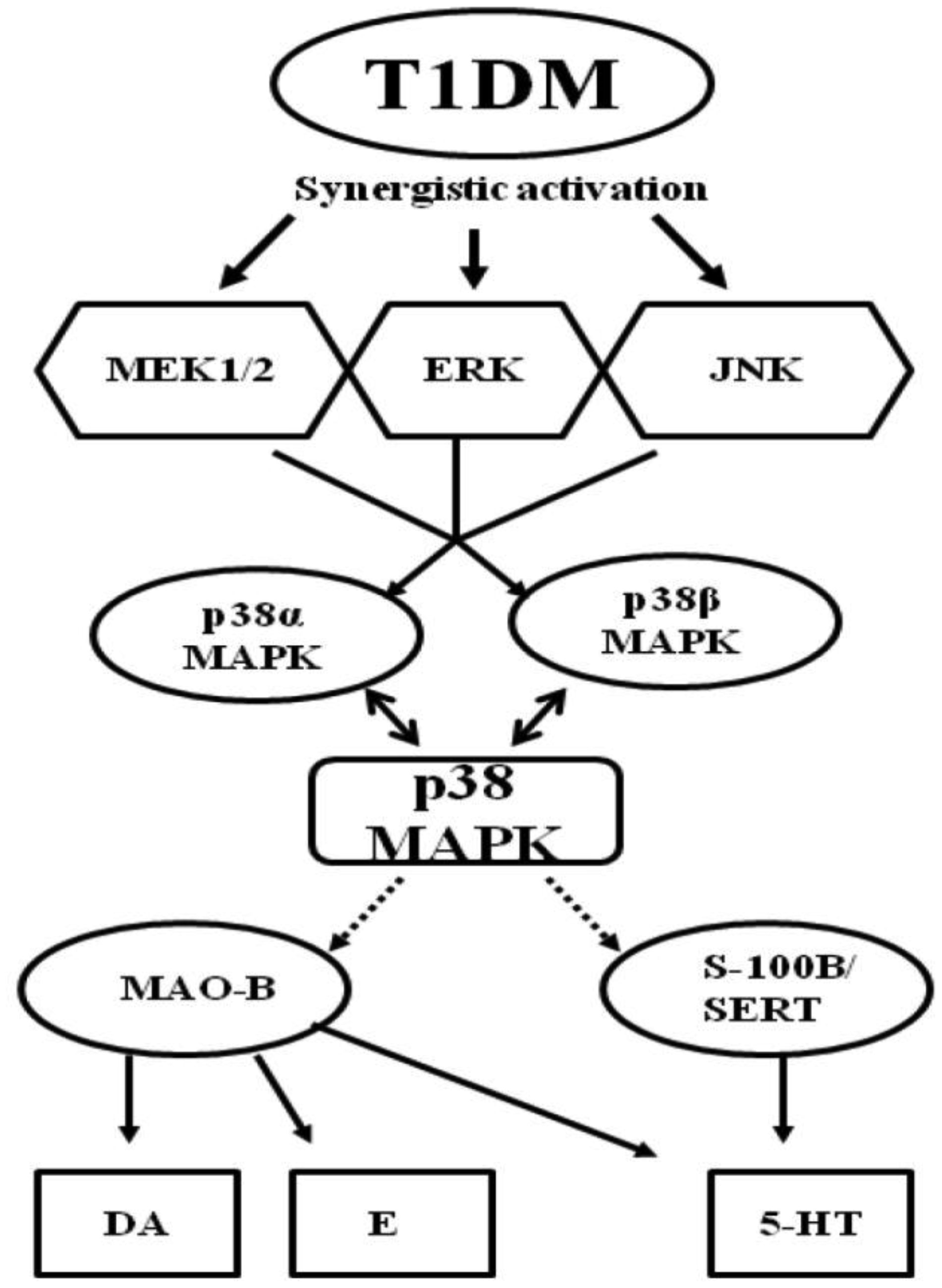
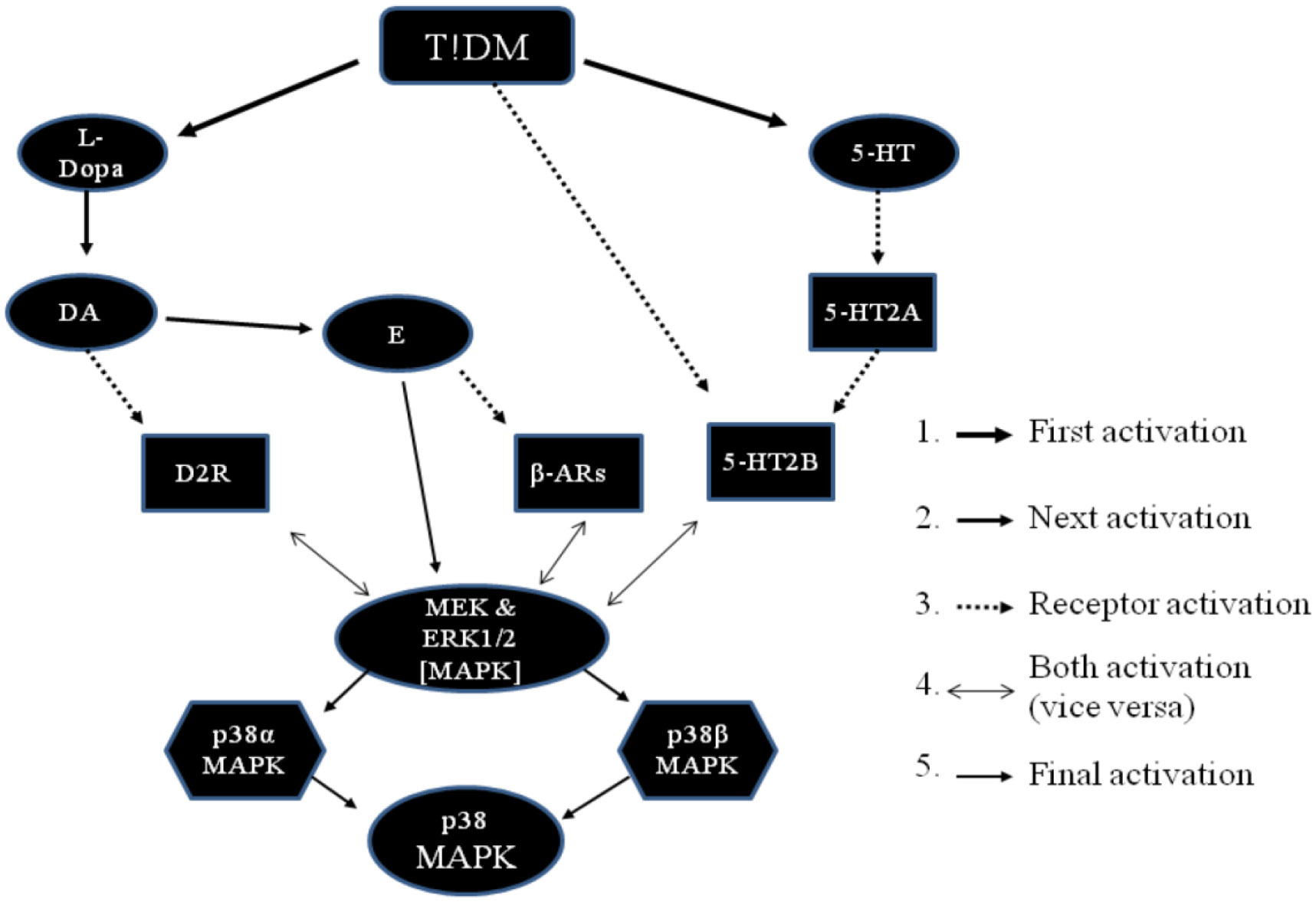
Figures(3) / Tables(1)

Ramakrishnan Ramugounder. The impact of p38 MAPK, 5-HT/DA/E signaling pathways in the development and progression of cardiovascular diseases and heart failure in type 1 diabetes[J]. AIMS Molecular Science, 2020, 7(4): 349-373. doi: 10.3934/molsci.2020017







 DownLoad:
DownLoad: