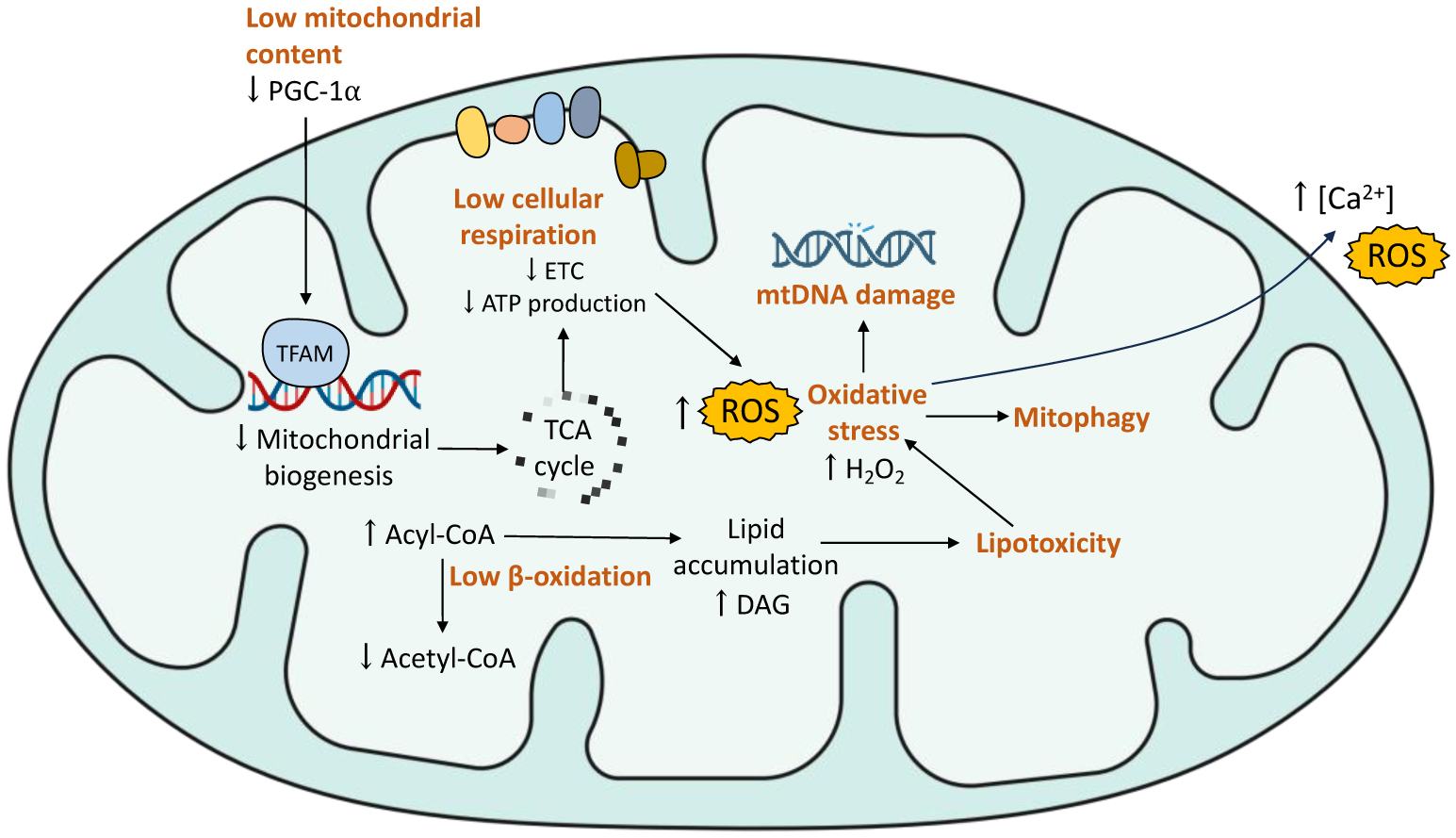
Type 2 diabetes (T2D) is characterized by a state of hyperglycemia in the blood due to insulin resistance developed by organs such as muscle, liver, and adipose tissue. A common factor in individuals with T2D is mitochondrial dysfunction. Mitochondria are dynamic organelles responsible for energy and antioxidant metabolism in the cells. Estrogens, such as 17β-estradiol (E2), are steroid hormones that have shown a great capacity to regulate mitochondrial function and dynamics through estrogen receptors (ERs), modulating the expression of mitochondrial biogenesis-related genes and cell signaling mechanisms. The accumulation of reactive oxygen species, the low capacity for ATP synthesis, and morphological alterations are some of the mitochondrial processes impaired in T2D. Insulin signaling and secretion by pancreatic β-cells, ATP-dependent processes, are also altered in T2D. In this review, mitochondria were exposed as the central axis for the action of estrogens in individuals with T2D. Estrogens increased glucose uptake, insulin signaling, and mitochondrial bioenergetics, and decreased ectopic lipid accumulation in non-adipose tissues and oxidative stress, among other processes, in various preclinical and clinical models of diabetes. The development of strategies to target compounds to mitochondria could represent a novel therapeutic alternative to potentiate the effects of estrogens on this organelle in patients with insulin resistance and T2D.
Citation: Geovanni Alberto Ruiz-Romero, Carolina Álvarez-Delgado. Effects of estrogens in mitochondria: An approach to type 2 diabetes[J]. AIMS Molecular Science, 2024, 11(1): 72-98. doi: 10.3934/molsci.2024006
Type 2 diabetes (T2D) is characterized by a state of hyperglycemia in the blood due to insulin resistance developed by organs such as muscle, liver, and adipose tissue. A common factor in individuals with T2D is mitochondrial dysfunction. Mitochondria are dynamic organelles responsible for energy and antioxidant metabolism in the cells. Estrogens, such as 17β-estradiol (E2), are steroid hormones that have shown a great capacity to regulate mitochondrial function and dynamics through estrogen receptors (ERs), modulating the expression of mitochondrial biogenesis-related genes and cell signaling mechanisms. The accumulation of reactive oxygen species, the low capacity for ATP synthesis, and morphological alterations are some of the mitochondrial processes impaired in T2D. Insulin signaling and secretion by pancreatic β-cells, ATP-dependent processes, are also altered in T2D. In this review, mitochondria were exposed as the central axis for the action of estrogens in individuals with T2D. Estrogens increased glucose uptake, insulin signaling, and mitochondrial bioenergetics, and decreased ectopic lipid accumulation in non-adipose tissues and oxidative stress, among other processes, in various preclinical and clinical models of diabetes. The development of strategies to target compounds to mitochondria could represent a novel therapeutic alternative to potentiate the effects of estrogens on this organelle in patients with insulin resistance and T2D.

| [1] |
DeFronzo RA, Ferrannini E, Groop L, et al. (2015) Type 2 diabetes mellitus. Nat Rev Dis Primers 1: 15019. https://doi.org/10.1038/nrdp.2015.19 
|
| [2] |
Ley SH, Hamdy O, Mohan V, et al. (2014) Prevention and management of type 2 diabetes: Dietary components and nutritional strategies. Lancet 383: 1999-2007. https://doi.org/10.1016/S0140-6736(14)60613-9
|
| [3] | Federation ID (2021) IDF diabetes atlas. Brussels, Belgium: International Diabetes Federation. Available from: https://diabetesatlas.org/. |
| [4] |
Pinti MV, Fink GK, Hathaway QA, et al. (2019) Mitochondrial dysfunction in type 2 diabetes mellitus: an organ-based analysis. Am J Physiol Endocrinol Metab 316: E268-E285. https://doi.org/10.1152/ajpendo.00314.2018
|
| [5] |
Duranova H, Valkova V, Knazicka Z, et al. (2020) Mitochondria: A worthwhile object for ultrastructural qualitative characterization and quantification of cells at physiological and pathophysiological states using conventional transmission electron microscopy. Acta Histochem 122: 151646. https://doi.org/10.1016/j.acthis.2020.151646
|
| [6] |
Spinelli JB, Haigis MC (2018) The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 20: 745-754. https://doi.org/10.1038/s41556-018-0124-1
|
| [7] |
Larsen S, Nielsen J, Hansen CN, et al. (2012) Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 590: 3349-3360. https://doi.org/10.1113/jphysiol.2012.230185
|
| [8] |
Lin CC, Cheng TL, Tsai WH, et al. (2012) Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep 2: 785. https://doi.org/10.1038/srep00785
|
| [9] |
Sazanov LA (2015) A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat Rev Mol Cell Biol 16: 375-388. https://doi.org/10.1038/nrm3997
|
| [10] |
Al Rasheed MRH, Tarjan G (2018) Succinate dehydrogenase complex: An updated review. Arch Pathol Lab Med 142: 1564-1570. https://doi.org/10.5858/arpa.2017-0285-RS
|
| [11] |
Neupane P, Bhuju S, Thapa N, et al. (2019) ATP synthase: Structure, function and inhibition. Biomol Concepts 10: 1-10. https://doi.org/10.1515/bmc-2019-0001
|
| [12] |
Prasun P (2020) Role of mitochondria in pathogenesis of type 2 diabetes mellitus. J Diabetes Metab Disord 19: 2017-2022. https://doi.org/10.1007/s40200-020-00679-x
|
| [13] |
Russell OM, Gorman GS, Lightowlers RN, et al. (2020) Mitochondrial diseases: Hope for the Future. Cell 181: 168-188. https://doi.org/10.1016/j.cell.2020.02.051
|
| [14] |
Miller WL, Auchus RJ (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 32: 81-151. https://doi.org/10.1210/er.2010-0013
|
| [15] |
Dard L, Blanchard W, Hubert C, et al. (2020) Mitochondrial functions and rare diseases. Mol Aspects Med 71: 100842. https://doi.org/10.1016/j.mam.2019.100842
|
| [16] |
Dong H, Tsai SY (2023) Mitochondrial properties in skeletal muscle fiber. Cells 12: 2183. https://doi.org/10.3390/cells12172183
|
| [17] |
Nunnari J, Suomalainen A (2012) Mitochondria: In sickness and in health. Cell 148: 1145-1159. https://doi.org/10.1016/j.cell.2012.02.035
|
| [18] |
Popov LD (2020) Mitochondrial biogenesis: An update. J Cell Mol Med 24: 4892-4899. https://doi.org/10.1111/jcmm.15194
|
| [19] |
Barshad G, Marom S, Cohen T, et al. (2018) Mitochondrial DNA transcription and its regulation: An evolutionary perspective. Trends Genet 34: 682-692. https://doi.org/10.1016/j.tig.2018.05.009
|
| [20] |
Wang F, Zhang D, Zhang D, et al. (2021) Mitochondrial protein translation: Emerging roles and clinical significance in disease. Front Cell Dev Biol 9: 675465. https://doi.org/10.3389/fcell.2021.675465
|
| [21] |
Demishtein-Zohary K, Azem A (2017) The TIM23 mitochondrial protein import complex: Function and dysfunction. Cell Tissue Res 367: 33-41. https://doi.org/10.1007/s00441-016-2486-7
|
| [22] |
Palikaras K, Lionaki E, Tavernarakis N (2015) Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ 22: 1399-1401. https://doi.org/10.1038/cdd.2015.86
|
| [23] |
Ding Q, Qi Y, Tsang SY (2021) Mitochondrial biogenesis, mitochondrial dynamics, and mitophagy in the maturation of cardiomyocytes. Cells 10: 2463. https://doi.org/10.3390/cells10092463
|
| [24] |
Zhang B, Pan C, Feng C, et al. (2022) Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox Rep 27: 45-52. https://doi.org/10.1080/13510002.2022.2046423
|
| [25] |
Schieber M, Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24: R453-R462. https://doi.org/10.1016/j.cub.2014.03.034
|
| [26] |
Cox AG, Winterbourn CC, Hampton MB (2009) Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem J 425: 313-325. https://doi.org/10.1042/BJ20091541
|
| [27] |
Ristow M, Schmeisser K (2014) Mitohormesis: Promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose Response 12: 288-341. https://doi.org/10.2203/dose-response.13-035.Ristow
|
| [28] |
Zarse K, Schmeisser S, Groth M, et al. (2012) Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab 15: 451-465. https://doi.org/10.1016/j.cmet.2012.02.013
|
| [29] |
Rovira-Llopis S, Banuls C, Diaz-Morales N, et al. (2017) Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol 11: 637-645. https://doi.org/10.1016/j.redox.2017.01.013
|
| [30] |
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605-610. https://doi.org/10.1038/nature07534
|
| [31] |
Tubbs E, Theurey P, Vial G, et al. (2014) Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63: 3279-3294. https://doi.org/10.2337/db13-1751
|
| [32] |
Head B, Griparic L, Amiri M, et al. (2009) Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol 187: 959-966. https://doi.org/10.1083/jcb.200906083
|
| [33] |
Consolato F, Maltecca F, Tulli S, et al. (2018) m-AAA and i-AAA complexes coordinate to regulate OMA1, the stress-activated supervisor of mitochondrial dynamics. J Cell Sci 131: jcs213546. https://doi.org/10.1242/jcs.213546
|
| [34] | Nan J, Nan C, Ye J, et al. (2019) EGCG protects cardiomyocytes against hypoxia-reperfusion injury through inhibition of OMA1 activation. J Cell Sci 132: jcs220871. https://doi.org/10.1242/jcs.220871 |
| [35] |
Otera H, Mihara K (2011) Molecular mechanisms and physiologic functions of mitochondrial dynamics. J Biochem 149: 241-251. https://doi.org/10.1093/jb/mvr002
|
| [36] |
Loson OC, Song Z, Chen H, et al. (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24: 659-667. https://doi.org/10.1091/mbc.E12-10-0721
|
| [37] |
Lackner LL (2014) Shaping the dynamic mitochondrial network. BMC Biol 12: 35. https://doi.org/10.1186/1741-7007-12-35
|
| [38] |
Denton RM (2009) Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787: 1309-1316. https://doi.org/10.1016/j.bbabio.2009.01.005
|
| [39] |
Gellerich FN, Gizatullina Z, Trumbeckaite S, et al. (2010) The regulation of OXPHOS by extramitochondrial calcium. Biochim Biophys Acta 1797: 1018-1027. https://doi.org/10.1016/j.bbabio.2010.02.005
|
| [40] |
Jana F, Bustos G, Rivas J, et al. (2019) Complex I and II are required for normal mitochondrial Ca2+ homeostasis. Mitochondrion 49: 73-82. https://doi.org/10.1016/j.mito.2019.07.004
|
| [41] |
Balderas E, Eberhardt DR, Lee S, et al. (2022) Mitochondrial calcium uniporter stabilization preserves energetic homeostasis during complex I impairment. Nat Commun 13: 2769. https://doi.org/10.1038/s41467-022-30236-4
|
| [42] |
Noyola-Martinez N, Halhali A, Barrera D (2019) Steroid hormones and pregnancy. Gynecol Endocrinol 35: 376-384. https://doi.org/10.1080/09513590.2018.1564742
|
| [43] |
Russell JK, Jones CK, Newhouse PA (2019) The role of estrogen in brain and cognitive aging. Neurotherapeutics 16: 649-665. https://doi.org/10.1007/s13311-019-00766-9
|
| [44] |
Soria-Jasso LE, Carino-Cortes R, Munoz-Perez VM, et al. (2019) Beneficial and deleterious effects of female sex hormones, oral contraceptives, and phytoestrogens by immunomodulation on the liver. Int J Mol Sci 20: 4694. https://doi.org/10.3390/ijms20194694
|
| [45] |
Bernasochi GB, Bell JR, Simpson ER, et al. (2019) Impact of estrogens on the regulation of white, beige, and brown adipose tissue depots. Compr Physiol 9: 457-475. https://doi.org/10.1002/cphy.c180009
|
| [46] |
Giatti S, Garcia-Segura LM, Barreto GE, et al. (2019) Neuroactive steroids, neurosteroidogenesis and sex. Prog Neurobiol 176: 1-17. https://doi.org/10.1016/j.pneurobio.2018.06.007
|
| [47] |
Zhang H, Cui D, Wang B, et al. (2007) Pharmacokinetic drug interactions involving 17alpha-ethinylestradiol: A new look at an old drug. Clin Pharmacokinet 46: 133-157. https://doi.org/10.2165/00003088-200746020-00003
|
| [48] |
Kuhl H (2005) Pharmacology of estrogens and progestogens: Influence of different routes of administration. Climacteric 8: 3-63. https://doi.org/10.1080/13697130500148875
|
| [49] |
Kumar R, Zakharov MN, Khan SH, et al. (2011) The dynamic structure of the estrogen receptor. J Amino Acids 2011: 812540. https://doi.org/10.4061/2011/812540
|
| [50] |
Gaudet HM, Cheng SB, Christensen EM, et al. (2015) The G-protein coupled estrogen receptor, GPER: The inside and inside-out story. Mol Cell Endocrinol 418: 207-219. https://doi.org/10.1016/j.mce.2015.07.016
|
| [51] |
Yasar P, Ayaz G, User SD, et al. (2017) Molecular mechanism of estrogen-estrogen receptor signaling. Reprod Med Biol 16: 4-20. https://doi.org/10.1002/rmb2.12006
|
| [52] |
Fuentes N, Silveyra P (2019) Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol 116: 135-170. https://doi.org/10.1016/bs.apcsb.2019.01.001
|
| [53] |
Li X, Zhang S, Safe S (2006) Activation of kinase pathways in MCF-7 cells by 17beta-estradiol and structurally diverse estrogenic compounds. J Steroid Biochem Mol Biol 98: 122-132. https://doi.org/10.1016/j.jsbmb.2005.08.018
|
| [54] |
Alvarez-Delgado C (2022) The role of mitochondria and mitochondrial hormone receptors on the bioenergetic adaptations to lactation. Mol Cell Endocrinol 551: 111661. https://doi.org/10.1016/j.mce.2022.111661
|
| [55] |
Chen JQ, Yager JD, Russo J (2005) Regulation of mitochondrial respiratory chain structure and function by estrogens/estrogen receptors and potential physiological/pathophysiological implications. Biochim Biophys Acta 1746: 1-17. https://doi.org/10.1016/j.bbamcr.2005.08.001
|
| [56] |
Escande A, Pillon A, Servant N, et al. (2006) Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem Pharmacol 71: 1459-1469. https://doi.org/10.1016/j.bcp.2006.02.002
|
| [57] |
Jeyakumar M, Carlson KE, Gunther JR, et al. (2011) Exploration of dimensions of estrogen potency: Parsing ligand binding and coactivator binding affinities. J Biol Chem 286: 12971-12982. https://doi.org/10.1074/jbc.M110.205112
|
| [58] |
Monje P, Boland R (2002) Expression and cellular localization of naturally occurring beta estrogen receptors in uterine and mammary cell lines. J Cell Biochem 86: 136-144. https://doi.org/10.1002/jcb.10193
|
| [59] |
Chen J, Li Y, Lavigne JA, et al. (1999) Increased mitochondrial superoxide production in rat liver mitochondria, rat hepatocytes, and HepG2 cells following ethinyl estradiol treatment. Toxicol Sci 51: 224-235. https://doi.org/10.1093/toxsci/51.2.224
|
| [60] |
Solakidi S, Psarra AMG, Sekeris CE (2005) Differential subcellular distribution of estrogen receptor isoforms: localization of ERalpha in the nucleoli and ERbeta in the mitochondria of human osteosarcoma SaOS-2 and hepatocarcinoma HepG2 cell lines. Biochim Biophys Acta 1745: 382-392. https://doi.org/10.1016/j.bbamcr.2005.05.010
|
| [61] |
Cammarata PR, Chu S, Moor A, et al. (2004) Subcellular distribution of native estrogen receptor alpha and beta subtypes in cultured human lens epithelial cells. Exp Eye Res 78: 861-871. https://doi.org/10.1016/j.exer.2003.09.027
|
| [62] |
Chen JQ, Delannoy M, Cooke C, et al. (2004) Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am J Physiol Endocrinol Metab 286: E1011-E1022. https://doi.org/10.1152/ajpendo.00508.2003
|
| [63] |
Chen JQ, Eshete M, Alworth WL, et al. (2004) Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J Cell Biochem 93: 358-373. https://doi.org/10.1002/jcb.20178
|
| [64] |
Chen JQ, Yager JD (2004) Estrogen's effects on mitochondrial gene expression: mechanisms and potential contributions to estrogen carcinogenesis. Ann N Y Acad Sci 1028: 258-272. https://doi.org/10.1196/annals.1322.030
|
| [65] |
Alvarez-Delgado C, Mendoza-Rodriguez CA, Picazo O, et al. (2010) Different expression of alpha and beta mitochondrial estrogen receptors in the aging rat brain: interaction with respiratory complex V. Exp Gerontol 45: 580-585. https://doi.org/10.1016/j.exger.2010.01.015
|
| [66] |
Chen JQ, Cammarata PR, Baines CP, et al. (2009) Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta 1793: 1540-1570. https://doi.org/10.1016/j.bbamcr.2009.06.001
|
| [67] |
Heine PA, Taylor JA, Iwamoto GA, et al. (2000) Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A 97: 12729-12734. https://doi.org/10.1073/pnas.97.23.12729
|
| [68] |
Musatov S, Chen W, Pfaff DW, et al. (2007) Silencing of estrogen receptor alpha in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci U S A 104: 2501-2506. https://doi.org/10.1073/pnas.0610787104
|
| [69] |
Gao Q, Horvath TL (2008) Cross-talk between estrogen and leptin signaling in the hypothalamus. Am J Physiol Endocrinol Metab 294: E817-E826. https://doi.org/10.1152/ajpendo.00733.2007
|
| [70] |
Foryst-Ludwig A, Clemenz M, Hohmann S, et al. (2008) Metabolic actions of estrogen receptor beta (ERbeta) are mediated by a negative cross-talk with PPARgamma. PLoS Genet 4: e1000108. https://doi.org/10.1371/journal.pgen.1000108
|
| [71] |
Torres MJ, Kew KA, Ryan TE, et al. (2018) 17beta-Estradiol directly lowers mitochondrial membrane microviscosity and improves bioenergetic function in skeletal muscle. Cell Metab 27: 167-179. https://doi.org/10.1016/j.cmet.2017.10.003
|
| [72] |
Torres MJ, Ryan TE, Lin CT, et al. (2018) Impact of 17beta-estradiol on complex I kinetics and H2O2 production in liver and skeletal muscle mitochondria. J Biol Chem 293: 16889-16898. https://doi.org/10.1074/jbc.RA118.005148
|
| [73] |
Moreno AJM, Moreira PI, Custodio JBA, et al. (2013) Mechanism of inhibition of mitochondrial ATP synthase by 17beta-estradiol. J Bioenerg Biomembr 45: 261-270. https://doi.org/10.1007/s10863-012-9497-1
|
| [74] |
Sastre J, Pallardo FV, Vina J (2003) The role of mitochondrial oxidative stress in aging. Free Radic Biol Med 35: 1-8. https://doi.org/10.1016/s0891-5849(03)00184-9
|
| [75] |
Borras C, Gambini J, Vina J (2007) Mitochondrial oxidant generation is involved in determining why females live longer than males. Front Biosci 12: 1008-1013. https://doi.org/10.2741/2120
|
| [76] |
Borras C, Gambini J, Gomez-Cabrera MC, et al. (2005) 17beta-oestradiol up-regulates longevity-related, antioxidant enzyme expression via the ERK1 and ERK2[MAPK]/NFkappaB cascade. Aging Cell 4: 113-118. https://doi.org/10.1111/j.1474-9726.2005.00151.x
|
| [77] |
Borras C, Gambini J, Lopez-Grueso R, et al. (2010) Direct antioxidant and protective effect of estradiol on isolated mitochondria. Biochim Biophys Acta 1802: 205-211. https://doi.org/10.1016/j.bbadis.2009.09.007
|
| [78] |
La Colla A, Vasconsuelo A, Boland R (2013) Estradiol exerts antiapoptotic effects in skeletal myoblasts via mitochondrial PTP and MnSOD. J Endocrinol 216: 331-341. https://doi.org/10.1530/JOE-12-0486
|
| [79] |
Bauza-Thorbrugge M, Galmés-Pascual BM, Sbert-Roig M, et al. (2017) Antioxidant peroxiredoxin 3 expression is regulated by 17beta-estradiol in rat white adipose tissue. J Steroid Biochem Mol Biol 172: 9-19. https://doi.org/10.1016/j.jsbmb.2017.05.008
|
| [80] |
Kalkhoran SB, Kararigas G (2022) Oestrogenic regulation of mitochondrial dynamics. Int J Mol Sci 23: 1118. https://doi.org/10.3390/ijms23031118
|
| [81] |
Tao Z, Cheng Z (2023) Hormonal regulation of metabolism-recent lessons learned from insulin and estrogen. Clin Sci (Lond) 137: 415-434. https://doi.org/10.1042/CS20210519
|
| [82] |
Sastre-Serra J, Nadal-Serrano M, Pons DG, et al. (2012) Mitochondrial dynamics is affected by 17beta-estradiol in the MCF-7 breast cancer cell line. Effects on fusion and fission related genes. Int J Biochem Cell Biol 44: 1901-1905. https://doi.org/10.1016/j.biocel.2012.07.012
|
| [83] |
Castracani CC, Longhitano L, Distefano A, et al. (2020) Role of 17beta-estradiol on cell proliferation and mitochondrial fitness in glioblastoma cells. J Oncol 2020: 2314693. https://doi.org/10.1155/2020/2314693
|
| [84] |
Satohisa S, Zhang HH, Feng L, et al. (2014) Endogenous NO upon estradiol-17beta stimulation and NO donor differentially regulate mitochondrial S-nitrosylation in endothelial cells. Endocrinology 155: 3005-3016. https://doi.org/10.1210/en.2013-2174
|
| [85] |
Zhou Z, Ribas V, Rajbhandari P, et al. (2018) Estrogen receptor α protects pancreatic beta-cells from apoptosis by preserving mitochondrial function and suppressing endoplasmic reticulum stress. J Biol Chem 293: 4735-4751. https://doi.org/10.1074/jbc.M117.805069
|
| [86] |
Lobaton CD, Vay L, Hernandez-Sanmiguel E, et al. (2005) Modulation of mitochondrial Ca2+ uptake by estrogen receptor agonists and antagonists. Br J Pharmacol 145: 862-871. https://doi.org/10.1038/sj.bjp.0706265
|
| [87] |
Vasan N, Toska E, Scaltriti M (2019) Overview of the relevance of PI3K pathway in HR-positive breast cancer. Ann Oncol 30: x3-x11. https://doi.org/10.1093/annonc/mdz281
|
| [88] |
Marchi S, Corricelli M, Branchini A, et al. (2019) Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca2+ levels and tumor growth. EMBO J 38: e99435. https://doi.org/10.15252/embj.201899435
|
| [89] |
Qin S, Yin J, Huang K (2016) Free fatty acids increase intracellular lipid accumulation and oxidative stress by modulating PPARalpha and SREBP-1c in L-02 cells. Lipids 51: 797-805. https://doi.org/10.1007/s11745-016-4160-y
|
| [90] |
Colak E, Pap D (2021) The role of oxidative stress in the development of obesity and obesity-related metabolic disorders. J Med Biochem 40: 1-9. https://doi.org/10.5937/jomb0-24652
|
| [91] |
Petersen MC, Shulman GI (2018) Mechanisms of insulin action and insulin resistance. Physiol Rev 98: 2133-2223. https://doi.org/10.1152/physrev.00063.2017
|
| [92] |
Kruger M, Kratchmarova I, Blagoev B, et al. (2008) Dissection of the insulin signaling pathway via quantitative phosphoproteomics. Proc Natl Acad Sci U S A 105: 2451-2456. https://doi.org/10.1073/pnas.0711713105
|
| [93] |
Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7: 85-96. https://doi.org/10.1038/nrm1837
|
| [94] |
Boura-Halfon S, Zick Y (2009) Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab 296: E581-E591. https://doi.org/10.1152/ajpendo.90437.2008
|
| [95] |
Petersen KF, Befroy D, Dufour S, et al. (2003) Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 300: 1140-1142. https://doi.org/10.1126/science.1082889
|
| [96] |
Kelley DE, Goodpaster B, Wing RR, et al. (1999) Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol 277: E1130-E1141. https://doi.org/10.1152/ajpendo.1999.277.6.E1130
|
| [97] |
Simoneau JA, Veerkamp JH, Turcotte LP, et al. (1999) Markers of capacity to utilize fatty acids in human skeletal muscle: relation to insulin resistance and obesity and effects of weight loss. FASEB J 13: 2051-2060. https://doi.org/10.1096/fasebj.13.14.2051
|
| [98] |
Kim JY, Hickner RC, Cortright RL, et al. (2000) Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab 279: E1039-E1044. https://doi.org/10.1152/ajpendo.2000.279.5.E1039
|
| [99] |
Szendroedi J, Phielix E, Roden M (2011) The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 8: 92-103. https://doi.org/10.1038/nrendo.2011.138
|
| [100] |
San-Millan I (2023) The key role of mitochondrial function in health and disease. Antioxidants (Basel) 12: 782. https://doi.org/10.3390/antiox12040782
|
| [101] |
Morino K, Petersen KF, Dufour S, et al. (2005) Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest 115: 3587-3593. https://doi.org/10.1172/JCI25151
|
| [102] |
Ritov VB, Menshikova EV, He J, et al. (2005) Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 54: 8-14. https://doi.org/10.2337/diabetes.54.1.8
|
| [103] |
Chomentowski P, Coen PM, Radikova Z, et al. (2011) Skeletal muscle mitochondria in insulin resistance: differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J Clin Endocrinol Metab 96: 494-503. https://doi.org/10.1210/jc.2010-0822
|
| [104] |
Amati F, Dube JJ, Alvarez-Carnero E, et al. (2011) Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: Another paradox in endurance-trained athletes?. Diabetes 60: 2588-2597. https://doi.org/10.2337/db10-1221
|
| [105] |
Bergman BC, Goodpaster BH (2020) Exercise and muscle lipid content, composition, and localization: Influence on muscle insulin sensitivity. Diabetes 69: 848-858. https://doi.org/10.2337/dbi18-0042
|
| [106] |
Sergi D, Naumovski N, Heilbronn LK, et al. (2019) Mitochondrial (dys)function and insulin resistance: From pathophysiological molecular mechanisms to the impact of diet. Front Physiol 10: 532. https://doi.org/10.3389/fphys.2019.00532
|
| [107] |
Holloszy JO (2009) Skeletal muscle “mitochondrial deficiency” does not mediate insulin resistance. Am J Clin Nutr 89: 463S-466S. https://doi.org/10.3945/ajcn.2008.26717C
|
| [108] |
Asmann YW, Stump CS, Short KR, et al. (2006) Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes 55: 3309-3319. https://doi.org/10.2337/db05-1230
|
| [109] |
Mogensen M, Sahlin K, Fernstrom M, et al. (2007) Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 56: 1592-1599. https://doi.org/10.2337/db06-0981
|
| [110] |
Phielix E, Schrauwen-Hinderling VB, Mensink M, et al. (2008) Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 57: 2943-2949. https://doi.org/10.2337/db08-0391
|
| [111] |
Koska J, Stefan N, Permana PA, et al. (2008) Increased fat accumulation in liver may link insulin resistance with subcutaneous abdominal adipocyte enlargement, visceral adiposity, and hypoadiponectinemia in obese individuals. Am J Clin Nutr 87: 295-302. https://doi.org/10.1093/ajcn/87.2.295
|
| [112] |
Petersen KF, Dufour S, Befroy D, et al. (2005) Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 54: 603-608. https://doi.org/10.2337/diabetes.54.3.603
|
| [113] |
Zhang D, Liu ZX, Choi CS, et al. (2007) Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci U S A 104: 17075-17080. https://doi.org/10.1073/pnas.0707060104
|
| [114] |
Zhang D, Christianson J, Liu ZX, et al. (2010) Resistance to high-fat diet-induced obesity and insulin resistance in mice with very long-chain acyl-CoA dehydrogenase deficiency. Cell Metab 11: 402-411. https://doi.org/10.1016/j.cmet.2010.03.012
|
| [115] |
Koliaki C, Szendroedi J, Kaul K, et al. (2015) Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab 21: 739-746. https://doi.org/10.1016/j.cmet.2015.04.004
|
| [116] |
Schmid AI, Szendroedi J, Chmelik M, et al. (2011) Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 34: 448-453. https://doi.org/10.2337/dc10-1076
|
| [117] |
Gancheva S, Kahl S, Pesta D, et al. (2022) Impaired hepatic mitochondrial capacity in nonalcoholic steatohepatitis associated with type 2 diabetes. Diabetes Care 45: 928-937. https://doi.org/10.2337/dc21-1758
|
| [118] |
De Pauw A, Tejerina S, Raes M, et al. (2009) Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. Am J Pathol 175: 927-939. https://doi.org/10.2353/ajpath.2009.081155
|
| [119] |
Lee JH, Park A, Oh KJ, et al. (2019) The role of adipose tissue mitochondria: Regulation of mitochondrial function for the treatment of metabolic diseases. Int J Mol Sci 20: 4924. https://doi.org/10.3390/ijms20194924
|
| [120] |
Chattopadhyay M, Guhathakurta I, Behera P, et al. (2011) Mitochondrial bioenergetics is not impaired in nonobese subjects with type 2 diabetes mellitus. Metabolism 60: 1702-1710. https://doi.org/10.1016/j.metabol.2011.04.015
|
| [121] |
Heinonen S, Buzkova J, Muniandy M, et al. (2015) Impaired mitochondrial biogenesis in adipose tissue in acquired obesity. Diabetes 64: 3135-3145. https://doi.org/10.2337/db14-1937
|
| [122] |
Bohm A, Keuper M, Meile T, et al. (2020) Increased mitochondrial respiration of adipocytes from metabolically unhealthy obese compared to healthy obese individuals. Sci Rep 10: 12407. https://doi.org/10.1038/s41598-020-69016-9
|
| [123] |
Meister BM, Hong SG, Shin J, et al. (2022) Healthy versus unhealthy adipose tissue expansion: The role of exercise. J Obes Metab Syndr 31: 37-50. https://doi.org/10.7570/jomes21096
|
| [124] |
Choo HJ, Kim JH, Kwon OB, et al. (2006) Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 49: 784-791. https://doi.org/10.1007/s00125-006-0170-2
|
| [125] |
Rong JX, Qiu Y, Hansen MK, et al. (2007) Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes 56: 1751-1760. https://doi.org/10.2337/db06-1135
|
| [126] |
Komatsu M, Takei M, Ishii H, et al. (2013) Glucose-stimulated insulin secretion: A newer perspective. J Diabetes Investig 4: 511-516. https://doi.org/10.1111/jdi.12094
|
| [127] |
Anello M, Lupi R, Spampinato D, et al. (2005) Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 48: 282-289. https://doi.org/10.1007/s00125-004-1627-9
|
| [128] |
Saxena R, de Bakker PI, Singer K, et al. (2006) Comprehensive association testing of common mitochondrial DNA variation in metabolic disease. Am J Hum Genet 79: 54-61. https://doi.org/10.1086/504926
|
| [129] |
Koeck T, Olsson AH, Nitert MD, et al. (2011) A common variant in TFB1M is associated with reduced insulin secretion and increased future risk of type 2 diabetes. Cell Metab 13: 80-91. https://doi.org/10.1016/j.cmet.2010.12.007
|
| [130] |
Sharoyko VV, Abels M, Sun J, et al. (2014) Loss of TFB1M results in mitochondrial dysfunction that leads to impaired insulin secretion and diabetes. Hum Mol Genet 23: 5733-5749. https://doi.org/10.1093/hmg/ddu288
|
| [131] |
Sidarala V, Pearson GL, Parekh VS, et al. (2020) Mitophagy protects β cells from inflammatory damage in diabetes. JCI Insight 5: e141138. https://doi.org/10.1172/jci.insight.141138
|
| [132] |
Vezza T, Diaz-Pozo P, Canet F, et al. (2022) The role of mitochondrial dynamic dysfunction in age-associated type 2 diabetes. World J Mens Health 40: 399-411. https://doi.org/10.5534/wjmh.210146
|
| [133] |
Shenouda SM, Widlansky ME, Chen K, et al. (2011) Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 124: 444-453. https://doi.org/10.1161/CIRCULATIONAHA.110.014506
|
| [134] |
Hu Q, Zhang H, Cortes NG, et al. (2020) Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ Res 126: 456-470. https://doi.org/10.1161/CIRCRESAHA.119.315252
|
| [135] |
Sidarala V, Zhu J, Levi-D'Ancona E, et al. (2022) Mitofusin 1 and 2 regulation of mitochondrial DNA content is a critical determinant of glucose homeostasis. Nat Commun 13: 2340. https://doi.org/10.1038/s41467-022-29945-7
|
| [136] |
Tubbs E, Chanon S, Robert M, et al. (2018) Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes 67: 636-650. https://doi.org/10.2337/db17-0316
|
| [137] |
Nieblas B, Perez-Trevino P, Garcia N (2022) Role of mitochondria-associated endoplasmic reticulum membranes in insulin sensitivity, energy metabolism, and contraction of skeletal muscle. Front Mol Biosci 9: 959844. https://doi.org/10.3389/fmolb.2022.959844
|
| [138] |
Dror V, Kalynyak TB, Bychkivska Y, et al. (2008) Glucose and endoplasmic reticulum calcium channels regulate HIF-1beta via presenilin in pancreatic beta-cells. J Biol Chem 283: 9909-9916. https://doi.org/10.1074/jbc.M710601200
|
| [139] |
Koval OM, Nguyen EK, Mittauer DJ, et al. (2023) Regulation of smooth muscle cell proliferation by mitochondrial Ca2+ in type 2 Diabetes. Int J Mol Sci 24: 12897. https://doi.org/10.3390/ijms241612897
|
| [140] |
Janssen I, Powell LH, Crawford S, et al. (2008) Menopause and the metabolic syndrome: The study of women's health across the nation. Arch Intern Med 168: 1568-1575. https://doi.org/10.1001/archinte.168.14.1568
|
| [141] |
Oya J, Nakagami T, Yamamoto Y, et al. (2014) Effects of age on insulin resistance and secretion in subjects without diabetes. Int Med 53: 941-947. https://doi.org/10.2169/internalmedicine.53.1580
|
| [142] |
Pu D, Tan R, Yu Q, et al. (2017) Metabolic syndrome in menopause and associated factors: A meta-analysis. Climacteric 20: 583-591. https://doi.org/10.1080/13697137.2017.1386649
|
| [143] |
Korljan B, Bagatin J, Kokic S, et al. (2010) The impact of hormone replacement therapy on metabolic syndrome components in perimenopausal women. Med Hypotheses 74: 162-163. https://doi.org/10.1016/j.mehy.2009.07.008
|
| [144] |
Lobo RA (2017) Hormone-replacement therapy: Current thinking. Nat Rev Endocrinol 13: 220-231. https://doi.org/10.1038/nrendo.2016.164
|
| [145] |
Bitoska I, Krstevska B, Milenkovic T, et al. (2016) Effects of hormone replacement therapy on insulin resistance in postmenopausal diabetic women. Open Access Maced J Med Sci 4: 83-88. https://doi.org/10.3889/oamjms.2016.024
|
| [146] |
Weigt C, Hertrampf T, Flenker U, et al. (2015) Effects of estradiol, estrogen receptor subtype-selective agonists and genistein on glucose metabolism in leptin resistant female Zucker diabetic fatty (ZDF) rats. J Steroid Biochem Mol Biol 154: 12-22. https://doi.org/10.1016/j.jsbmb.2015.06.002
|
| [147] |
Ribas V, Drew BG, Zhou Z, et al. (2016) Skeletal muscle action of estrogen receptor alpha is critical for the maintenance of mitochondrial function and metabolic homeostasis in females. Sci Transl Med 8: 334ra354. https://doi.org/10.1126/scitranslmed.aad3815
|
| [148] |
Diaz A, Lopez-Grueso R, Gambini J, et al. (2019) Sex differences in age-associated type 2 diabetes in rats-role of estrogens and oxidative stress. Oxid Med Cell Longev 2019: 6734836. https://doi.org/10.1155/2019/6734836
|
| [149] |
Le May C, Chu K, Hu M, et al. (2006) Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci U S A 103: 9232-9237. https://doi.org/10.1073/pnas.0602956103
|
| [150] |
Alonso-Magdalena P, Ropero AB, Carrera MP, et al. (2008) Pancreatic insulin content regulation by the estrogen receptor ER alpha. PLoS One 3: e2069. https://doi.org/10.1371/journal.pone.0002069
|
| [151] |
Kilic G, Alvarez-Mercado AI, Zarrouki B, et al. (2014) The islet estrogen receptor-α is induced by hyperglycemia and protects against oxidative stress-induced insulin-deficient diabetes. PLoS One 9: e87941. https://doi.org/10.1371/journal.pone.0087941
|
| [152] |
Liu S, Le May C, Wong WPS, et al. (2009) Importance of extranuclear estrogen receptor-alpha and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes 58: 2292-2302. https://doi.org/10.2337/db09-0257
|
| [153] |
Hevener A, Reichart D, Janez A, et al. (2002) Female rats do not exhibit free fatty acid-induced insulin resistance. Diabetes 51: 1907-1912. https://doi.org/10.2337/diabetes.51.6.1907
|
| [154] |
Camporez JP, Lyu K, Goldberg EL, et al. (2019) Anti-inflammatory effects of oestrogen mediate the sexual dimorphic response to lipid-induced insulin resistance. J Physiol 597: 3885-3903. https://doi.org/10.1113/JP277270
|
| [155] |
Gonzalez-Granillo M, Savva C, Li X, et al. (2019) ERβ activation in obesity improves whole body metabolism via adipose tissue function and enhanced mitochondria biogenesis. Mol Cell Endocrinol 479: 147-158. https://doi.org/10.1016/j.mce.2018.10.007
|
| [156] |
Galmes-Pascual BM, Martinez-Cignoni MR, Moran-Costoya A, et al. (2020) 17β-estradiol ameliorates lipotoxicity-induced hepatic mitochondrial oxidative stress and insulin resistance. Free Radic Biol Med 150: 148-160. https://doi.org/10.1016/j.freeradbiomed.2020.02.016
|
| [157] |
Nauck MA, Vardarli I, Deacon CF, et al. (2011) Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: What is up, what is down?. Diabetologia 54: 10-18. https://doi.org/10.1007/s00125-010-1896-4
|
| [158] |
Drucker DJ, Nauck MA (2006) The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 368: 1696-1705. https://doi.org/10.1016/S0140-6736(06)69705-5
|
| [159] |
Buteau J (2008) GLP-1 receptor signaling: effects on pancreatic beta-cell proliferation and survival. Diabetes Metab 34: S73-S77. https://doi.org/10.1016/S1262-3636(08)73398-6
|
| [160] |
Tiano JP, Tate CR, Yang BS, et al. (2015) Effect of targeted estrogen delivery using glucagon-like peptide-1 on insulin secretion, insulin sensitivity and glucose homeostasis. Sci Rep 5: 10211. https://doi.org/10.1038/srep10211
|
| [161] |
Fuselier T, de Sa PM, Qadir MMF, et al. (2022) Efficacy of glucagon-like peptide-1 and estrogen dual agonist in pancreatic islets protection and pre-clinical models of insulin-deficient diabetes. Cell Rep Med 3: 100598. https://doi.org/10.1016/j.xcrm.2022.100598
|
| [162] |
Finan B, Yang B, Ottaway N, et al. (2012) Targeted estrogen delivery reverses the metabolic syndrome. Nat Med 18: 1847-1856. https://doi.org/10.1038/nm.3009
|
| [163] | Jiang Q, Yin J, Chen J, et al. (2020) Mitochondria-targeted antioxidants: A step towards disease treatment. Oxid Med Cell Longev 2020: 8837893. https://doi.org/10.1155/2020/8837893 |
| [164] |
Zielonka J, Joseph J, Sikora A, et al. (2017) Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev 117: 10043-10120. https://doi.org/10.1021/acs.chemrev.7b00042
|
| [165] |
Cheng G, Zielonka J, McAllister DM, et al. (2013) Mitochondria-targeted vitamin E analogs inhibit breast cancer cell energy metabolism and promote cell death. BMC Cancer 13: 285. https://doi.org/10.1186/1471-2407-13-285
|
| [166] |
Kelso GF, Porteous CM, Coulter CV, et al. (2001) Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem 276: 4588-4596. https://doi.org/10.1074/jbc.M009093200
|

Figures(2)

Geovanni Alberto Ruiz-Romero, Carolina Álvarez-Delgado. Effects of estrogens in mitochondria: An approach to type 2 diabetes[J]. AIMS Molecular Science, 2024, 11(1): 72-98. doi: 10.3934/molsci.2024006







 DownLoad:
DownLoad: