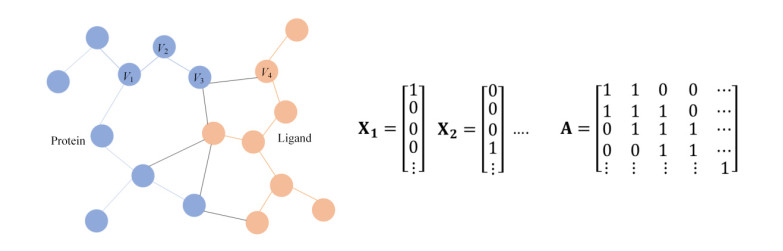
Estimating the binding affinity between proteins and drugs is very important in the application of structure-based drug design. Currently, applying machine learning to build the protein-ligand binding affinity prediction model, which is helpful to improve the performance of classical scoring functions, has attracted many scientists' attention. In this paper, we have developed an affinity prediction model called GAT-Score based on graph attention network (GAT). The protein-ligand complex is represented by a graph structure, and the atoms of protein and ligand are treated in the same manner. Two improvements are made to the original graph attention network. Firstly, a dynamic feature mechanism is designed to enable the model to deal with bond features. Secondly, a virtual super node is introduced to aggregate node-level features into graph-level features, so that the model can be used in the graph-level regression problems. PDBbind database v.2018 is used to train the model. Finally, the performance of GAT-Score was tested by the scheme $C_s$ (Core set as the test set) and CV (Cross-Validation). It has been found that our results are better than most methods from machine learning models with traditional molecular descriptors.
Citation: Hong Yuan, Jing Huang, Jin Li. Protein-ligand binding affinity prediction model based on graph attention network[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 9148-9162. doi: 10.3934/mbe.2021451
Estimating the binding affinity between proteins and drugs is very important in the application of structure-based drug design. Currently, applying machine learning to build the protein-ligand binding affinity prediction model, which is helpful to improve the performance of classical scoring functions, has attracted many scientists' attention. In this paper, we have developed an affinity prediction model called GAT-Score based on graph attention network (GAT). The protein-ligand complex is represented by a graph structure, and the atoms of protein and ligand are treated in the same manner. Two improvements are made to the original graph attention network. Firstly, a dynamic feature mechanism is designed to enable the model to deal with bond features. Secondly, a virtual super node is introduced to aggregate node-level features into graph-level features, so that the model can be used in the graph-level regression problems. PDBbind database v.2018 is used to train the model. Finally, the performance of GAT-Score was tested by the scheme $C_s$ (Core set as the test set) and CV (Cross-Validation). It has been found that our results are better than most methods from machine learning models with traditional molecular descriptors.

| [1] |
M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray, R. D. Taylor, Improved protein-ligand docking using GOLD, Proteins, 52 (2003), 609-623. doi: 10.1002/prot.10465

|
| [2] |
R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, et al., Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy, J. Med. Chem., 47 (2004), 1739-1749. doi: 10.1021/jm0306430
|
| [3] |
W. Zhe, H. Sun, X. Yao, L. Dan, T. Hou, Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: the prediction accuracy of sampling power and scoring power, Phys. Chem. Chem. Phys., 18 (2016), 1-27. doi: 10.1039/C6CP90001A
|
| [4] |
Z. Gaieb, S. Liu, S. Gathiaka, M. Chiu, H. Yang, C. Shao, et al., D3R Grand Challenge 2: blind prediction of protein-ligand poses, affinity rankings, and relative binding free energies, J. Comput. Aid. Mol. Des., 32 (2018), 1-20. doi: 10.1007/s10822-017-0088-4
|
| [5] | H. Li, K. S. Leung, M. H. Wong, P. J. Ballester, Improving AutoDock Vina using random forest: the growing accuracy of binding affinity prediction by the effective exploitation of larger data sets, Mol. Biol., 34 (2015), 115-126. |
| [6] |
G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew, et al., Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function, J. Comput. Chem., 19 (1998), 1639-1662. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B
|
| [7] |
V. Y. Tanchuk, V. O. Tanin, A. I. Vovk, G. Poda, A new, improved hybrid scoring function for molecular docking and scoring based on AutoDock and AutoDock Vina, Chem. Biol. Drug Des., 87 (2016), 618-625. doi: 10.1111/cbdd.12697
|
| [8] | O. Trott, A. J. Olson, Software news and update AutoDock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 31 (2010), 455-461. |
| [9] | A. A. Toropov, A. P. Toropova, R. G. Diaza, E. Benfenati, G. Gini, SMILES-based optimal descriptors: QSAR modeling of estrogen receptor binding affinity by correlation balance, Struct. Chem., 23 (2011), 529-544. |
| [10] | M. Wójcikowski, M. Kukiełka, M. M. Stepniewska-Dziubinska, P. Siedlecki, Development of a protein-ligand extended connectivity (PLEC) fingerprint and its application for binding affinity predictions, Bioinformatics, 35 (2018), 1334-1344. |
| [11] |
C. Zixuan, G. W. Wei, R. L. Dunbrack, TopologyNet: Topology based deep convolutional and multi-task neural networks for biomolecular property predictions, PLoS Comput. Biol., 13 (2017), e1005690. doi: 10.1371/journal.pcbi.1005690
|
| [12] | H. Zhai, Research on image recognition based on deep learning technology, in International Conference on Advanced Materials and Information Technology Processing, (2016), 266-270. |
| [13] |
B. J. Abbaschian, D. Sierra-Sosa, A. Elmaghraby, Deep learning techniques for speech emotion recognition, from databases to models, Sensors, 21 (2021), 1249. doi: 10.3390/s21041249
|
| [14] | P. Klosowski, Deep learning for natural language processing and language modelling, in 2018 Signal Processing: Algorithms, Architectures, Arrangements, and Applications, (2018), 223-228. |
| [15] |
M. M. Stepniewska-dziubinska, P. Zielenkiewicz, P. Siedlecki, Development and evaluation of a deep learning model for protein-ligand binding affinity prediction, Bioinformatics, 34 (2018), 3666-3674. doi: 10.1093/bioinformatics/bty374
|
| [16] | Y. Li, M. A. Rezaei, C. Li, X. Li, D. Wu, DeepAtom: A framework for protein-ligand binding affinity prediction, in IEEE International Conference on Bioinformatics and Biomedicine, (2019), 303-310. |
| [17] |
M. Ragoza, J. Hochuli, E. Idrobo, J. Sunseri, D. R. Koes, Protein-ligand scoring with convolutional neural networks, J. Chem. Inf. Model., 57 (2017), 942-957. doi: 10.1021/acs.jcim.6b00740
|
| [18] | I. Wallach, M. Dzamba, A. Heifets, AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery, preprint, arXiv: 1510.02855. |
| [19] |
D. Mishkin, N. Sergievskiy, J. Matas, Systematic evaluation of convolution neural network advances on the Imagenet, Comput. Vis. Image Und., 161 (2017), 11-19. doi: 10.1016/j.cviu.2017.05.007
|
| [20] | Z. Wu, S. Pan, F. Chen, G. Long, C. Zhang, P. S. Yu, A comprehensive survey on graph neural networks, IEEE Trans. Neural Netw. Learn. Syst., 32 (2019), 4-24. |
| [21] | J. Zhou, G. Cui, S. Hu, Z. Zhang, C. Yang, Z. Liu, et al., Graph neural networks: A review of methods and applications, AI Open, 1 (2021), 57-81. |
| [22] |
S. Zhang, H. Tong, J. Xu, R. Maciejewski, Graph convolutional networks: a comprehensive review, Comput. Soc. Netw., 6 (2019), 1-23. doi: 10.1186/s40649-019-0061-6
|
| [23] | D. Bahdanau, K. Cho, Y. Bengio, Neural machine translation by jointly learning to align and translate, in International Conference on Learning Representations, 2015. |
| [24] | J. Cheng, L. Dong, M. Lapata, Long short-term memory-networks for machine reading, in Proceedings of the 2016 Conference on Empirical Methods in Natural Language Processing, (2016), 551-561. |
| [25] | P. Veličković, G. Cucurull, A. Casanova, A. Romero, P. Liò, Y. Bengio, Graph attention networks, preprint, arXiv: 1710.10903. |
| [26] |
M. Segler, T. Kogej, C. Tyrchan, M. P. Waller, Generating focused molecule libraries for drug discovery with recurrent neural networks, ACS Cent. Sci., 4 (2018), 120-131. doi: 10.1021/acscentsci.7b00512
|
| [27] |
Y. Li, M. Su, Z. Liu, J. Li, J. Liu, L. Han, et al., Assessing protein-ligand interaction scoring functions with the CASF-2013 benchmark, Nat. Protoc., 13 (2018), 666-680. doi: 10.1038/nprot.2017.114
|
| [28] |
F. Scarselli, M. Gori, A. C. Tsoi, M. Hagenbuchner, G. Monfardini, The graph neural network model, IEEE Trans. Neural Netw., 20 (2009), 61-80. doi: 10.1109/TNN.2008.2005605
|
| [29] | RDKit, Available from: http://www.rdkit.org/. |
| [30] | K. Liu, X. Sun, L. Jia, J. Ma, H. Xing, J. Wu, et al., Chemi-net: a graph convolutional network for accurate drug property prediction, Int. J. Mol. Sci., 20 (2018), 3389. |
| [31] | S. Ioffe, C. Szegedy, Batch normalization: Acceleration deep network training by reducing internal covariate shift, in International Conference on Machine Learning, (2015), 448-456. |
| [32] |
M. A. Hossam, R. M. Nihar, Task-specific scoring functions for predicting ligand binding poses and affinity and for screening enrichment, J. Chem. Inf. Model, 58 (2018), 119-132. doi: 10.1021/acs.jcim.7b00309
|
| [33] | H. Ashtawy, N. Mahapatra, A comparative assessment of predictive accuracies of conventional and machine learning scoring functions for protein-ligand binding affinity prediction, IEEE ACM Trans. Comput. Biol. Bioinf., 12 (2010), 335-347. |
| [34] |
Y. Li, L. Han, Z. Liu, R. Wang, Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results, J. Chem. Inf. Model., 54 (2014), 1717-1736. doi: 10.1021/ci500081m
|
| [35] |
T. Cheng, X. Li, Y. Li, Z. Liu, R. Wang, Comparative assessment of scoring functions on a diverse test set, J. Chem. Inf. Model., 49 (2009), 1079-1093. doi: 10.1021/ci9000053
|
| [36] | H. M. Ashtawy, N. R. Mahapatra, BgN-Score and BsN-Score: Bagging and boosting based ensemble neural networks scoring functions for accurate binding affinity prediction of protein-ligand complexes, BMC Bioinf., 16 (2015), 1-12. |
| [37] | H. M. Ashtawy, N. R. Mahapatra, Machine-learning scoring functions for identifying native poses of ligands docked to known and novel proteins, BMC Bioinf., 16 (2015), S3. |
| [38] | Z. Meng, K. Xia, Persistent spectral-based machine learning (PerSpect ML) for protein-ligand binding affinity prediction, Sci. Adv., 7 (2021), eabc5329. |
| [39] | M. Su, Q. Yang, Y. Du, G. Feng, Z. Liu, Y. Li, et al., Comparative assessment of scoring functions: the CASF-2016 update, J. Chem. Inf. Model., 59 (2018), 895-913. |
| [40] | RCSB PDB, Available from: http://www.rcsb.org/. |






Figures(7) / Tables(4)

Hong Yuan, Jing Huang, Jin Li. Protein-ligand binding affinity prediction model based on graph attention network[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 9148-9162. doi: 10.3934/mbe.2021451







 DownLoad:
DownLoad: