Citation: Alhaji Cherif, Vaibhav Maheshwari, Doris Fuertinger, Gudrun Schappacher-Tilp, Priscila Preciado, David Bushinsky, Stephan Thijssen, Peter Kotanko. A mathematical model of the four cardinal acid-base disorders[J]. Mathematical Biosciences and Engineering, 2020, 17(5): 4457-4476. doi: 10.3934/mbe.2020246

| [1] | W. B. Busa, R. Nuccitelli, Metabolic regulation via intracellular pH, Am. J. Physiol., 246 (1984), R409-R438. |
| [2] | T. E. DeCoursey, The intimate and controversial relationship between voltage-gated proton channels and the phagocyte NADPH oxidase, Immunol. Rev., 273 (2016), 194-218. |
| [3] | E. K. Hoffmann, L. O. Simonsen, Membrane mechanisms in volume and pH regulation in vertebrate cells, Physiol. Rev., 69 (1989), 315-382. |
| [4] | A. Roos, W. F. Boron, Intracellular pH, Physiol. Rev., 61 (1981), 296-434. |
| [5] | A. Schonichen, B. A. Webb, M. P. Jacobson, D. L. Barber, Considering protonation as a posttranslational modification regulating protein structure and function, Annu. Rev. Biophys., 42 (2013), 289-314. |
| [6] | K. F. Atkinson, S. M. Nauli, pH sensors and ion Transporters: Potential therapeutic targets for acid-base disorders, Int. J. Pharma Res. Rev., 5 (2016), 51. |
| [7] | W. F. Boron, Acid-base transport by the renal proximal tubule, J. Am. Soc. Nephrol., 17 (2006), 2368-2382. |
| [8] | V. Fencl, J. Vale, J. Broch, Respiration and cerebral blood flow in metabolic acidosis and alkalosis in humans, J. Appl. Phys., 27 (1969), 67-76 |
| [9] | L. L. Hamm, N. Nakhou, K. S. Hering-Smith, Acid-base homeostasis, Clin. J. Am. Soc. Nephrol., 10 (2015), 2232-2242. |
| [10] | D. Hornick, An approach to the analysis of arterial blood gases and acid-base disorders, Virtual Hospital, University of Iowa Health Care [On-line], 2003. |
| [11] | M. Levitzky, Pulmonary Physiology, McGraw-Hill Book Company, 2003. |
| [12] | C. Lote, Principles of Renal Physiology, Kluwer Academic Publishers, 1999. |
| [13] | N. A. Masco, Acid-base homeostasis, J. Infusion Nurs., 39 (2016), 288-295. |
| [14] | R. Mitchell, M. Singer, Respiration and cerebrospinal fluid pH in metabolic acidosis and alkalosis, J. Appl. Phys., 20 (1965), 905-911. |
| [15] | G. T. Nagami, L. L. Hamm, Regulation of acid-base balance in chronic kidney disease, Adv. Chronic kidney Dis., 24 (2017), 274-279. |
| [16] | R. Pitts, Physiology of the Kidney and Body Fluids, Year Book Medical Publishers Inc, 1970. |
| [17] | J. Poppell, P. Vanamee, K. Roberts, H. Randall, The effect of ventilatory insufficiency on respiratory compensations in metabolic acidosis and alkalosis, J. Lab. Clin. Med., 47 (1956), 885-890. |
| [18] | R. Quigley, Acid-base homeostasis. Clin. Pediatr. Nephrol., 2016, 235. |
| [19] | W. B. Schwartz, J. J. Cohen, The nature of the renal response to chronic disorders of acid-base equilibrium, Am. J. Med., 64 (1978), 417-428. |
| [20] | L. A. Skelton, W. F. Boron, Y. Zhou, Acid-base transport by the renal proximal tubule, J. Nephrol., 23 (2010), S4. |
| [21] | J. Lemann Jr., D. A. Bushinsky, L. L. Hamm, Bone buffering of acid and base in humans, Am. J. Physiol. Renal Physiol., 285 (2003), F811-F832. |
| [22] | H. Davenport, The ABC of acid-base chemistry, The University of Chicago, Chicago, IL, 1974. |
| [23] | R. Hainsworth, Acid-Base Balance, Manchester University, UK, 1986. |
| [24] | J. B. West, Respiratory Physiology, 9th edition, Lippincott Williams and Wilkins, Philadelphia, PA, 2012. |
| [25] | J. Widdicombe, A. Davies, Respiratory Physiology, Edward Arnold Publishers, 1983. |
| [26] | W. Lang, R. Zander, Prediction of dilutional acidosis based on the revised classical dilution concept for bicarbonate, J. Appl. Phys., 98 (2005), 62-71. |
| [27] | M. B. Wolf, E. C. DeLand, A mathematical model of blood-interstitial acid-base balance: Application to dilution acidosis and acid-base status, J. Appl. Phys., 110 ( 2011), 988-1002. |
| [28] | K. Annan, Mathematical modeling of the dynamic exchange of solutes during bicarbonate dialysis, Math. Comput. Modell., 55 (2012), 1691-1704. |
| [29] | L. Coli, M. Ursino, A. De Pascalis, C. Brighenti, V. Dalmastri, G. La Manna, et al., Evaluation of intradialytic solute and fluid kinetics, Blood Purif., 18 (2000), 37-49. |
| [30] | R. K. Dash, J. B. Bassingthwaighte, Simultaneous bloodtissue exchange of oxygen, carbon dioxide, bicarbonate, and hydrogen ion, Ann. Biomed. Eng., 34 (2006), 1129-1148. |
| [31] | S. Marano, M. Marano, Frontiers in hemodialysis: Solutions and implications of mathematical models for bicarbonate restoring, Biomed. Signal Process. Control, 52 (2019), 321-329. |
| [32] | N. K. Martin, E. A. Gaffney, R. A. Gatenby, R. J. Gillies, I. F. Robey, P. K. Maini, A mathematical model of tumour and blood pHe regulation: The HCO3-/CO2 buffering system, Math. Biosci., 230 (2011), 1-11. |
| [33] | J. A. Sargent, M. Marano, S. Marano, F. J. Gennari, Acidbase homeostasis during hemodialysis: New insights into the mystery of bicarbonate disappearance during treatment, Semin. Dial., (2018), 1-11. |
| [34] | O. Thews, H. Hutten, A comprehensive model of the dynamic exchange processes during hemodialysis, Med. Prog. Technol., 16 (1990), 145-161. |
| [35] | M. Ursino, L. Coli, C. Brighenti, L. Chiari, A. De Pascalis, G. Avanzolini, Prediction of solute kinetics, acid-base status, and blood volume changes during profiled hemodialysis, Ann. Biomed. Eng., 28 (2000), 204-216. |
| [36] | K. A. Hasselbalch, Die berechnung der wasserstoffzahl des blutes aus der freien und gebundenen kohlensure desselben, und die sauerstoffbindung des blutes als funktion der wasserstoffzahl, Biochem. Z., 78 (1917), 112-144. |
| [37] | L. J. Henderson, Concerning the relationship between the strength of acids and their capacity to preserve neutrality, Am. J. Physiol., 21 (1908), 173-179. |
| [38] | C. Chegwidden, E. Edwards, Carbonic Anhydrases: New Horizons, Birkhauser Press, 2000. |
| [39] | J. J. Batzel, F. Kappel, D. Schneditz, H. T. Tran, Cardiovascular and Respiratory Systems: Modeling, Analysis, and Control, SIAM, Philadelphia, 2007. |
| [40] | Memorang, Compensation Reactions to Acid/Base Imbalance [online]. Available from: https://www.memorangapp.com/flashcards/94371/Compensation+Reactions+to+Acid/2FBase+Imbalance (last accessed 24 April 2020). |
| [41] | M. S. Albert, R. B. Dell, R. W. Winters, Quantitative displacement of acid-base equilibrium in metabolic acidosis, Ann. Int. Med., 66 (1967), 312-322. |
| [42] | D. A. Bushinsky, F. L. Coe, C. Katzenberg, J. P. Szidon, J. H. Parks, Arterial PCO2 in chronic metabolic acidosis, Kidney Int., 22 (1982), 311-314. |
| [43] | K. Engel, R. B. Dell, W. J. Rahill, C. R. Denning, R. W. Winters, Quantitative displacement of acid-base equilibrium in chronic respiratory acidosis, J. Appl. Phys., 24 (1968), 288-295. |
| [44] | S. B. Gonzlez, G. Menga, G. A. Raimondi, H. Tighiouart, H. J. Adrogu, N. E. Madias, Secondary response to chronic respiratory acidosis in humans: A prospective study, Kidney Int. Rep., 3 (2018), 1163-1170. |
| [45] | A. Hasan, The Analysis of Blood Gases: Handbook of Blood Gas/Acid-Base Interpretation, Springer London, (2013), 253-266. |
| [46] | S. Javaheri, N. S. Shore, B. Rose, H. Kazemi, Compensatory hypoventilation in metabolic alkalosis, Chest, 81 (1982), 296-301. |
| [47] | K. Roberts, J. Poppell, P. Vanamee, R. Beals, H. Randall, Evaluation of respiratory compensation in metabolic alkalosis, J. Clin. Invest., 35 (1956), 261-266. |
| [48] | S. M. Blower, H. Dowlatabadi, Sensitivity and uncertainty analysis of complex models of disease transmission: An HIV model, as an example, Int. Stat. Rev., 2 (1994), 229-243. |
| [49] | S. Marino, I. B. Hogue, C. J. Ray, D. E. Kirschner, A methodology for performing global uncertainty and sensitivity analysis in systems biology, J. Theor. Biol., 254 (2008), 178-196 |
| [50] | D. A. Bushinsky, T. Hostetter, G. Klaerner, Y. Stasiv, C. Lockey, S. McNulty, et al., Randomized, controlled trial of tRC101 to increase serum bicarbonate in patients with CKD, Clin. J. Am. Soc. Nephrol., 13 (2018), 26-35. |
| [51] | R. C. De Sousa, J. T. Harrington, E. S. Ricanati, J. W. Shelkrot, W. B. Schwartz, Renal regulation of acid-base equilibrium during chronic administration of mineral acid, J. Clin. Invest., 53 (1974), 465-476. |
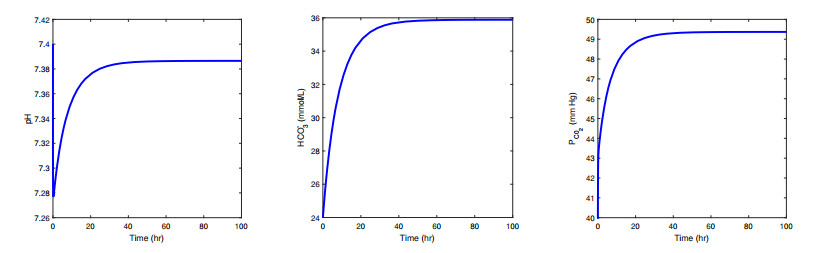
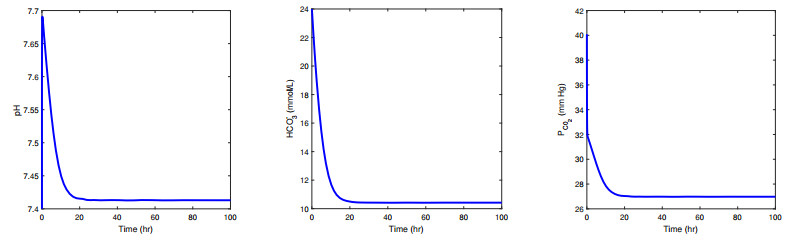
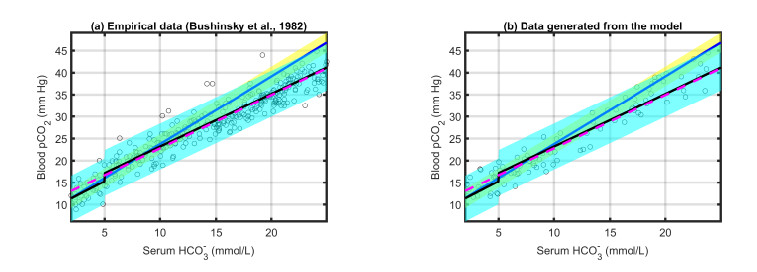
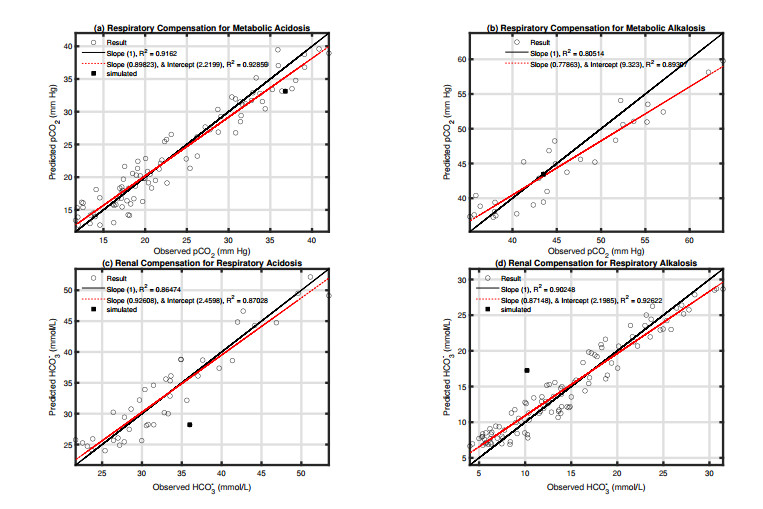
Figures(10) / Tables(3)

Alhaji Cherif, Vaibhav Maheshwari, Doris Fuertinger, Gudrun Schappacher-Tilp, Priscila Preciado, David Bushinsky, Stephan Thijssen, Peter Kotanko. A mathematical model of the four cardinal acid-base disorders[J]. Mathematical Biosciences and Engineering, 2020, 17(5): 4457-4476. doi: 10.3934/mbe.2020246







 DownLoad:
DownLoad: