Citation: Sebastien FM Chastin, Juliet A Harvey, Philippa M Dall, Lianne McInally, Alexandra Mavroeidi, Dawn A Skelton. Beyond “#endpjparalysis”, tackling sedentary behaviour in health care[J]. AIMS Medical Science, 2019, 6(1): 67-75. doi: 10.3934/medsci.2019.1.67

| [1] | Baldwin C, van Kessel G, Phillips A, et al. (2017) Accelerometry shows inpatients with acute medical or surgical conditions spend little time upright and are highly sedentary: Systematic review. Phys Ther 97: 1044–1065. |
| [2] |
Tremblay MS, Aubert S, Barnes JD, et al. (2017) Sedentary Behavior Research Network (SBRN) - Terminology Consensus Project process and outcome. Int J Behav Nutr Phys Act 14: 75. doi: 10.1186/s12966-017-0525-8

|
| [3] |
Chastin SFM, Scwartz U, Skelton DA (2013) Development of a consensus taxonomy of sedentary behaviors (SIT): Report of Delphi Round 1. PLoS One 8: e82313. doi: 10.1371/journal.pone.0082313
|
| [4] |
Grant PM, Granat MH, Thow MK, et al. (2010) Analyzing free-living physical activity of older adults in different environments using body-worn activity monitors. J Aging Phys Act 18: 171–184. doi: 10.1123/japa.18.2.171
|
| [5] | Sjöholm A, Skarin M, Churilov L, et al. (2014) Sedentary behaviour and physical activity of people with stroke in rehabilitation hospitals. Stroke Res Treat 2014: 591897. |
| [6] |
Karbiener M, Pisani DF, Frontini A, et al. (2014) MicroRNA-26 family is required for human adipogenesis and drives characteristics of brown adipocytes. Stem Cells 32: 1578–1590. doi: 10.1002/stem.1603
|
| [7] |
Taraldsen K, Thingstad P, Sletvold O, et al. (2015) The long-term effect of being treated in a geriatric ward compared to an orthopaedic ward on six measures of free-living physical behavior 4 and 12 months after a hip fracture - A randomised controlled trial. BMC Geriatr 15: 160. doi: 10.1186/s12877-015-0153-6
|
| [8] |
Egerton T, Maxwell DJ, Granat MH (2006) Mobility activity of stroke patients during inpatient rehabilitation. Hong Kong Physiother J 24: 8–15. doi: 10.1016/S1013-7025(07)70003-9
|
| [9] |
Harvey JA, Chastin SFM, Skelton DA (2018) What happened to my legs when I broke my arm? Aims Med Sci 5: 252–258. doi: 10.3934/medsci.2018.3.252
|
| [10] |
Chastin SFM, Egerton T, Leask C, et al. (2015) Meta-analysis of the relationship between breaks in sedentary behavior and cardiometabolic health. Obesity 23: 1800–1810. doi: 10.1002/oby.21180
|
| [11] |
Matthews CE, Keadle SK, Troiano RP, et al. (2016) Accelerometer-measured dose-response for physical activity, sedentary time, and mortality in US adults. Am J Clin Nutr 104: 1424–1432. doi: 10.3945/ajcn.116.135129
|
| [12] |
Biswas A, Oh PI, Faulkner GE, et al. (2015) Sedentary time and its association with risk for disease incidence, mortality, and hospitalization in adults: A systematic review and meta-analysis. Ann Intern Med 162: 123–132. doi: 10.7326/M14-1651
|
| [13] |
Ekegren CL, Beck B, Climie RE, et al. (2018) Physical activity and sedentary behavior subsequent to serious orthopedic injury: A systematic review. Arch Phys Med Rehabil 99: 164–177. doi: 10.1016/j.apmr.2017.05.014
|
| [14] |
Zusman EZ, Dawes MG, Edwards N, et al. (2018) A systematic review of evidence for older adults' sedentary behavior and physical activity after hip fracture. Clin Rehabil 32: 679–691. doi: 10.1177/0269215517741665
|
| [15] |
Tieges Z, Mead G, Allerhand M, et al. (2015) Sedentary behavior in the first year after stroke: A longitudinal cohort study with objective measures. Arch Phys Med Rehabil 96: 15–23. doi: 10.1016/j.apmr.2014.08.015
|
| [16] |
Kronborg L, Bandholm T, Palm H, et al. (2016) Physical activity in the acute ward following hip fracture surgery is associated with less fear of falling. J Aging Phys Act 24: 525–532. doi: 10.1123/japa.2015-0071
|
| [17] |
Krumholz HM (2013) Post-hospital syndrome-an acquired, transient condition of generalized risk. N Engl J Med 368: 100–102. doi: 10.1056/NEJMp1212324
|
| [18] |
Hodgson CL, Capell E, Tipping CJ (2018) Early mobilization of patients in intensive care: Organization, communication and safety factors that influence translation into clinical practice. Crit Care 22: 77. doi: 10.1186/s13054-018-1998-9
|
| [19] |
Dogra S, Ashe MC, Biddle SJH, et al. (2017) Sedentary time in older men and women: an international consensus statement and research priorities. Br J Sports Med 51: 1526–1532. doi: 10.1136/bjsports-2016-097209
|
| [20] |
Kortebein P (2009) Rehabilitation for hospital-associated deconditioning. Am J Phys Med Rehabil 88: 66–77. doi: 10.1097/PHM.0b013e3181838f70
|
| [21] | Skelton D, Van Wijck F, Grant M, et al. (2014) Are physiotherapists contributing to patient harm? Are your patients FIT TO SIT? Agility 20. |
| [22] | Boltz M, Resnick B, Capezuti E, et al. (2014) Activity restriction vs. self-direction: hospitalised older adults' response to fear of falling. Int J Older People Nurs 9: 44–53. |
| [23] |
Resnick B, Galik E, Wells CL, et al. (2015) Optimizing physical activity among older adults post trauma: Overcoming system and patient challenges. Int J Orthop Trauma Nurs 19: 194–206. doi: 10.1016/j.ijotn.2015.03.007
|
| [24] |
Owen N, Sugiyama T, Eakin EE, et al. (2011) Adults' sedentary behavior determinants and interventions. Am J Prev Med 41: 189–196. doi: 10.1016/j.amepre.2011.05.013
|
| [25] |
Chastin SFM, De Craemer M, Lien N, et al. (2016) The SOS-framework (Systems of Sedentary behaviours): An international transdisciplinary consensus framework for the study of determinants, research priorities and policy on sedentary behaviour across the life course: A DEDIPAC-study. Int J Behav Nutr Phys Act 13: 83. doi: 10.1186/s12966-016-0409-3
|
| [26] | Organisation for Economic and Cooperation Development (2009) Applications of Complexity Science for Public Policy. |
| [27] |
Finegood DT, Karanfil O, Matteson CL (2008) Getting from analysis to action: framing obesity research, policy and practice with a solution-oriented complex systems lens. Healthc Pap 9: 36–41. doi: 10.12927/hcpap.2008.20184
|
| [28] |
Greenhalgh T, Jackson C, Shaw S, et al. (2016) Achieving research impact through co-creation in community-based health services: Literature review and case study. Milbank Q 94: 392–429. doi: 10.1111/1468-0009.12197
|
| [29] |
Boltz M, Resnick B, Capezuti E, et al. (2012) Functional decline in hospitalized older adults: Can nursing make a difference? Geriatr Nurs 33: 272–279. doi: 10.1016/j.gerinurse.2012.01.008
|
| [30] | Langley GJ, Moen R, Nolan KM, et al. (2009) The improvement guide: A practical approach to enhancing organizational performance. 2nd Edition. |
| [31] | Hartley PJ, Keevil VL, Alushi L, et al. (2017) Earlier physical therapy input is associated with a reduced length of hospital stay and reduced care needs on discharge in frail older inpatients: An observational study. J Geriatr Phys Ther doi: 10.1519/JPT.0000000000000134. |
| [32] |
Dall P, Coulter EH, Fitzsimons C, et al. (2017) The TAxonomy of Self-reported Sedentary behaviour Tools (TASST) framework for development, comparison and evaluation of self-report tools: Content analysis and systematic review. BMJ Open 7: e013844. doi: 10.1136/bmjopen-2016-013844
|
| [33] |
Chastin SFM, Dontje ML, Skelton DA, et al. (2018) Systematic comparative validation of self-report measures of sedentary time against an objective measure of postural sitting (activPAL). Int J Behav Nutr Phys Act 15: 21. doi: 10.1186/s12966-018-0652-x
|
| [34] |
Crouter SE, Clowers KG, Bassett DR Jr (2006) A novel method for using accelerometer data to predict energy expenditure. J Appl Physiol 100: 1324–1331. doi: 10.1152/japplphysiol.00818.2005
|
| [35] |
Sellers C, Dall P, Grant M, et al. (2016) Validity and reliability of the activPAL3 for measuring posture and stepping in adults and young people. Gait Posture 43: 42–47. doi: 10.1016/j.gaitpost.2015.10.020
|
| [36] |
Fortune E, Lugade VA, Amin S, et al. (2015) Step detection using multi- versus single tri-axial accelerometer-based systems. Physiol Meas 36: 2519–2535. doi: 10.1088/0967-3334/36/12/2519
|
| [37] |
Cook DJ, Thompson JE, Prinsen SK, et al. (2013) Functional recovery in the elderly after major surgery: Assessment of mobility recovery using wireless technology. Ann Thorac Surg 96: 1057–1061. doi: 10.1016/j.athoracsur.2013.05.092
|
| [38] |
Ryan CG, Grant PM, Tigbe WW, et al. (2006) The validity and reliability of a novel activity monitor as a measure of walking. Br J Sports Med 40: 779–784. doi: 10.1136/bjsm.2006.027276
|
| [39] |
Loveday A, Sherar LB, Sanders JP, et al. (2015) Technologies that assess the location of physical activity and sedentary behavior: A systematic review. J Med Internet Res 17: e192. doi: 10.2196/jmir.4761
|
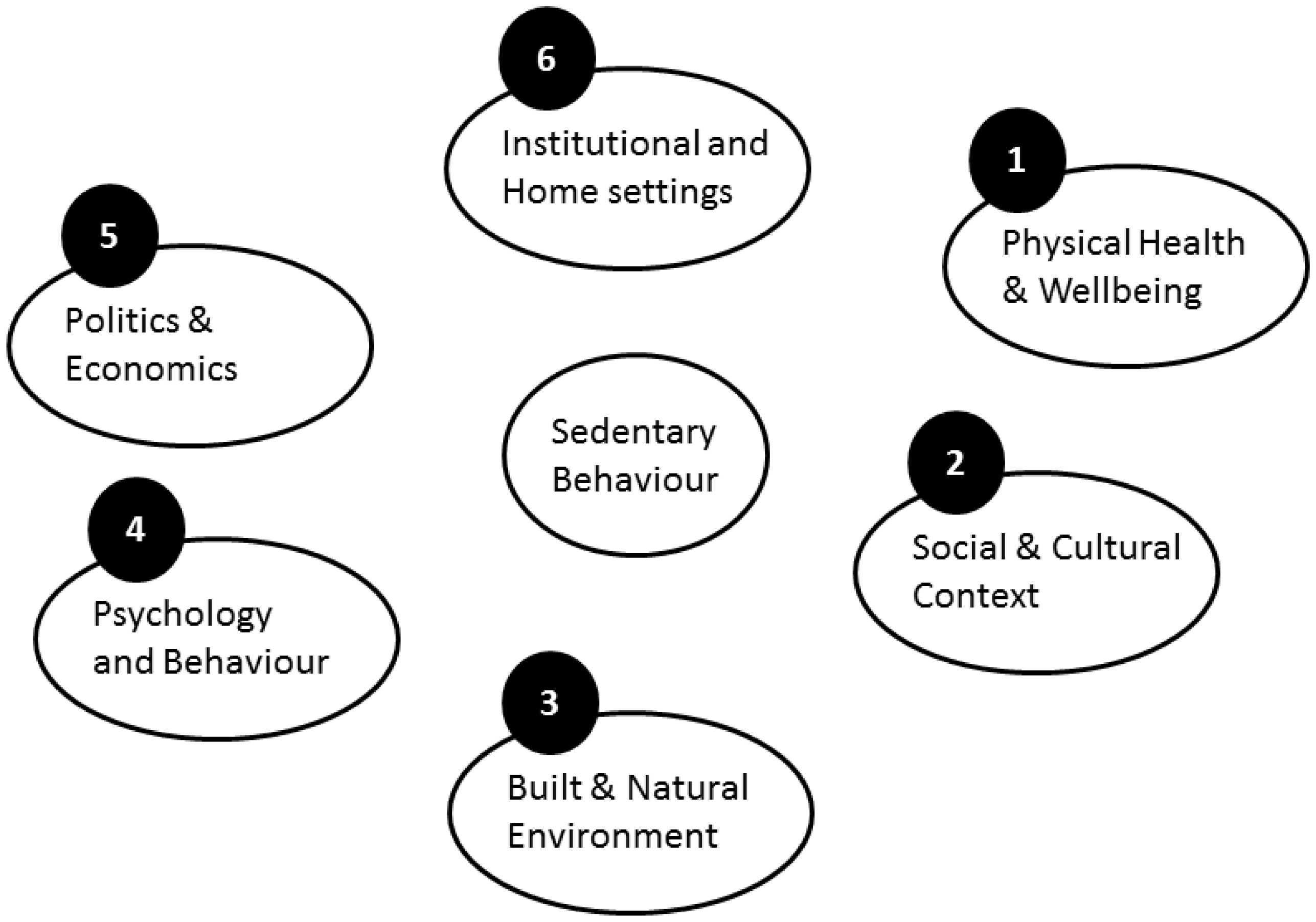
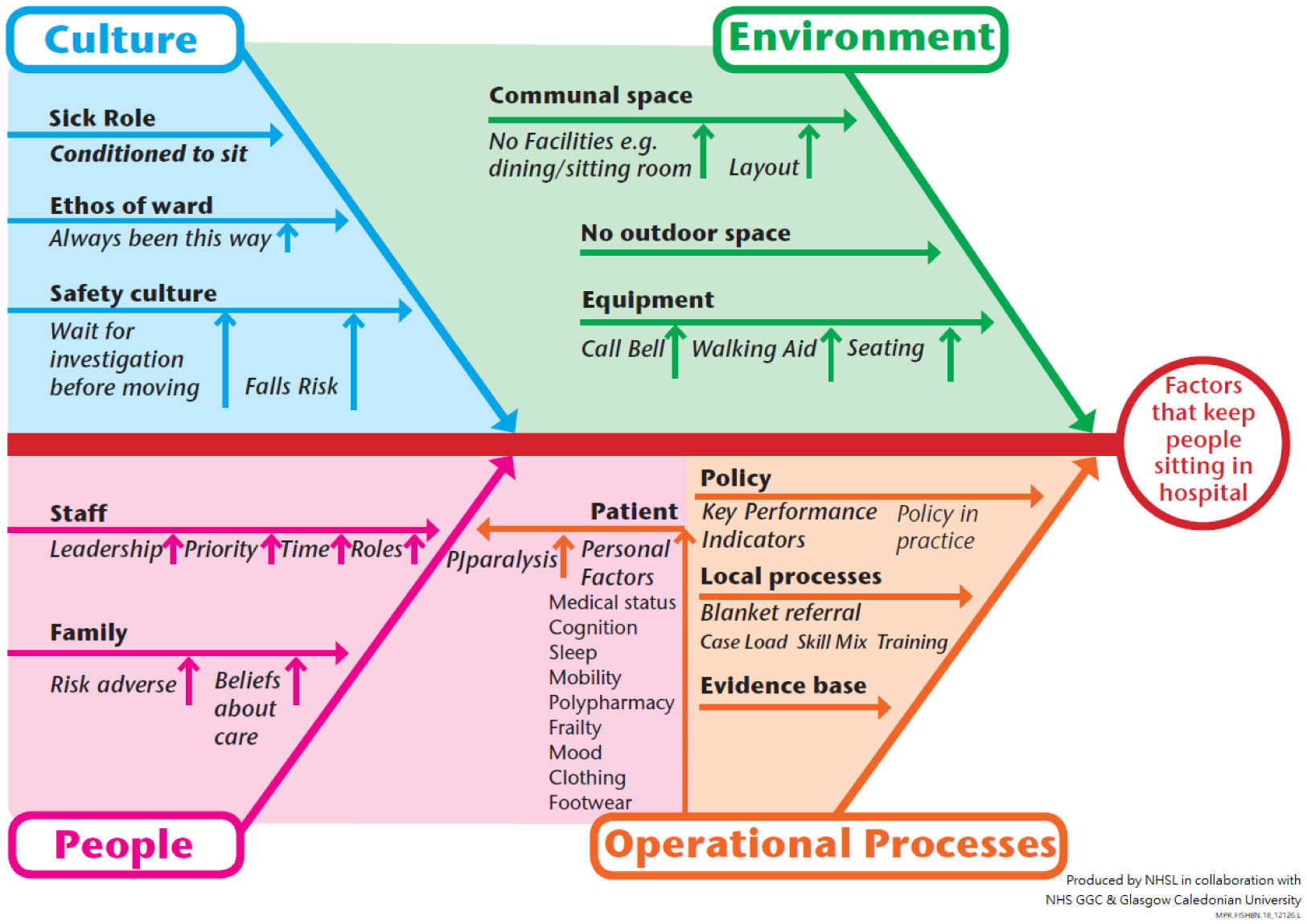
Figures(2)

Sebastien FM Chastin, Juliet A Harvey, Philippa M Dall, Lianne McInally, Alexandra Mavroeidi, Dawn A Skelton. Beyond “#endpjparalysis”, tackling sedentary behaviour in health care[J]. AIMS Medical Science, 2019, 6(1): 67-75. doi: 10.3934/medsci.2019.1.67







 DownLoad:
DownLoad: