Citation: Yunxiang Zhang, Lvcheng Jin, Bo Wang, Dehong Hu, Leqiang Wang, Pan Li, Junling Zhang, Kai Han, Geng Tian, Dawei Yuan, Jialiang Yang, Wei Tan, Xiaoming Xing, Jidong Lang. DL-CNV: A deep learning method for identifying copy number variations based on next generation target sequencing[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 202-215. doi: 10.3934/mbe.2020011

| [1] | S. A. McCarroll and D. M. Altshuler, Copy-number variation and association studies of human disease, Nat. Genet., 39(2007), S37-42. |
| [2] | P. Liu, C. M. Carvalho, P. J. Hastings, et al., Mechanisms for recurrent and complex human genomic rearrangements, Curr. Opin. Genet. Dev., 22(2012), 211-220. |
| [3] |
A. P. de Koning, W. Gu, T. A. Castoe, et al., Repetitive elements may comprise over two-thirds of the human genome, Plos Genet.,7 (2011), e1002384. doi: 10.1371/journal.pgen.1002384

|
| [4] |
M. Zarrei, J. R. MacDonald, D. Merico, et al., A copy number variation map of the human genome, Nat. Rev. Genet., 16(2015), 172-183. doi: 10.1038/nrg3871
|
| [5] | J. L. Freeman, G. H. Perry, L. Feuk, et al., Copy number variation: new insights in genome diversity, Genome Res., 16(2006), 949-961. |
| [6] | S. F. Chin, A. E. Teschendorff, J. C. Marioni, et al., High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer, Genome Biol., 8(2007), R215. |
| [7] | D. He, N. Furlotte and E. Eskin, Detection and reconstruction of tandemly organized de novo copy number variations, BMC Bioinf., 11(2010), S12. |
| [8] |
G. Klambauer, K. Schwarzbauer, A. Mayr, et al., cn.MOPS: Mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate, Nucleic Acids Res., 40 (2012), e69. doi: 10.1093/nar/gks003
|
| [9] | E. Talevich, A. H. Shain, T. Botton, et al., CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing, PLoS Comput. Biol., 12(2016), e1004873. |
| [10] | A. Abyzov, A. E. Urban, M. Snyder, et al., CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing, Genome Res., 21(2011), 974-984. |
| [11] | V. Boeva, T. Popova, K. Bleakley, et al., Control-FREEC: Atool for assessing copy number and allelic content using next-generation sequencing data, Bioinf., 28(2012), 423-425. |
| [12] | G. Onsongo, L. B. Baughn, M. Bower, et al., CNV-RF Is a Random Forest-Based Copy Number Variation Detection Method Using Next-Generation Sequencing, J. Mol. Diagn., 18(2016), 872-881. |
| [13] | C. Wang, J. M. Evans, A. V. Bhagwate, et al., PatternCNV: Aversatile tool for detecting copy number changes from exome sequencing data, Bioinf., 30(2014), 2678-2680. |
| [14] | J. Budczies, N. Pfarr, A. Stenzinger, et al., Ioncopy: Anovel method for calling copy number alterations in amplicon sequencing data including significance assessment, Oncotarget, 7 (2016), 13236-13247. |
| [15] | Y. L. Cun, L. Bottou, Y. Bengio, et al., Gradient-Based Learning Applied to Document Recognition, Proc. IEEE., 86 (1998), 2278-2324. |
| [16] | J. Zhou and O. G. Troyanskaya, Predicting effects of noncoding variants with deep learning-based sequence model, Nat. Methods,12 (2015), 931-934. |
| [17] | R. Poplin, D. Newburger, J. Dijamco, et al., Creating a universal SNP and small indel variant caller with deep neural networks, BioRxiv, (2016), 092890. |
| [18] | M. X. Sliwkowski, J. A. Lofgren, G. D. Lewis, et al., Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin), Semin Oncol., 26(1999), 60-70. |
| [19] | S. Ahn, M. Hong, M. Van Vrancken, et al., A nCounter CNV Assay to Detect HER2 Amplification: A Correlation Study with Immunohistochemistry and In Situ Hybridization in Advanced Gastric Cancer, Mol. Diagn. Ther., 20(2016), 375-383. |
| [20] | F. Sircoulomb, I. Bekhouche, P. Finetti, et al., Genome profiling of ERBB2-amplified breast cancers, BMC Cancer,10 (2010), 539. |
| [21] | S. Kim, T. M. Kim, D. W. Kim, et al., Acquired Resistance of MET-Amplified Non-small Cell Lung Cancer Cells to the MET Inhibitor Capmatinib, Cancer Res. Treat., 5 (2019), 951-962. |
| [22] | N. Pfarr, R. Penzel, F. Klauschen, et al., Copy number changes of clinically actionable genes in melanoma, non-small cell lung cancer and colorectal cancer-A survey across 822 routine diagnostic cases, Genes Chromosomes Cancer, 55 (2016), 821-833. |
| [23] | F. Zare, M. Dow, N. Monteleone, et al., An evaluation of copy number variation detection tools for cancer using whole exome sequencing data, BMC Bioinf., 18(2017), 286. |
| [24] | H. Li and R. Durbin, Fast and accurate short read alignment with Burrows-Wheeler transform, Bioinf., 25(2009), 1754-1760. |
| [25] | H. Li, B. Handsaker, A. Wysoker, et al., The Sequence Alignment/Map format and SAMtools, Bioinf., 25(2009), 2078-2079. |
| [26] | M. Abadi, P. Barham, J. Chen, et al., TensorFlow: A system for large-scale machine learning, 12th Symposium on Operating Systems Design and Implementation (OSDI), 2016, 265-283. Available from: https://www.usenix.org/conference/osdi16/technical-sessions/presentation/abadi. |
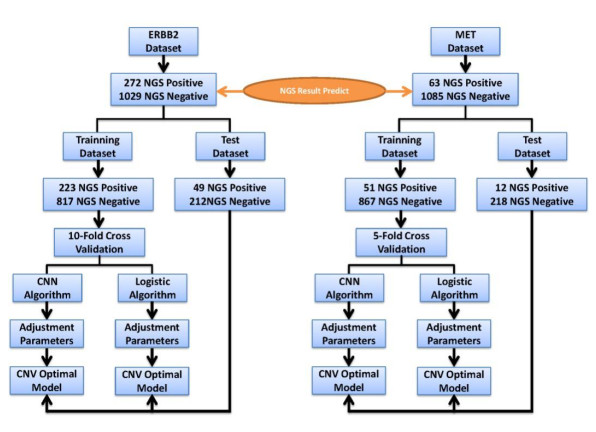
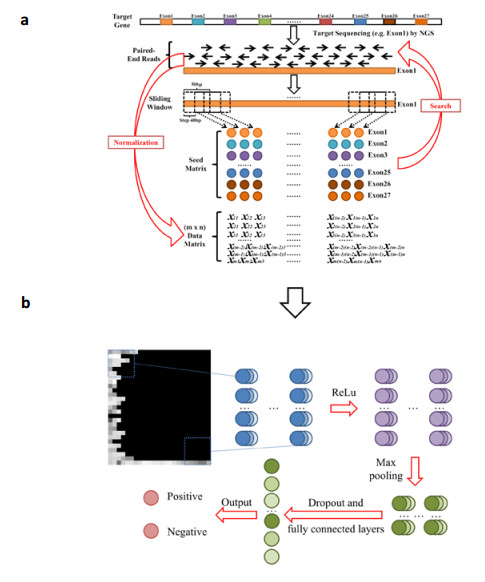
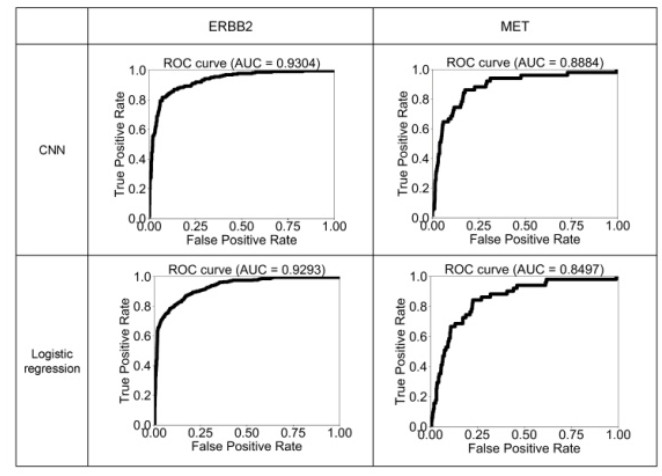
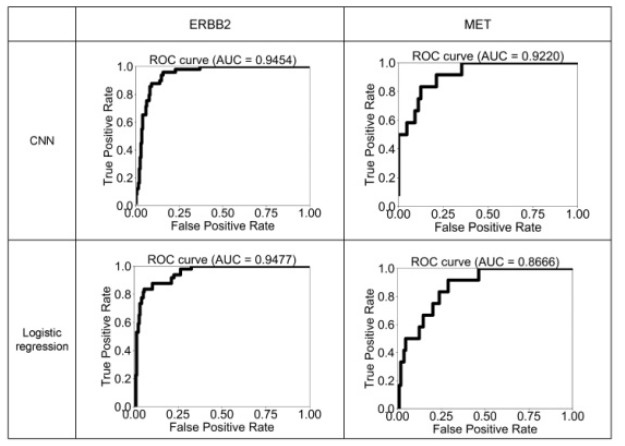
Figures(6) / Tables(2)

Yunxiang Zhang, Lvcheng Jin, Bo Wang, Dehong Hu, Leqiang Wang, Pan Li, Junling Zhang, Kai Han, Geng Tian, Dawei Yuan, Jialiang Yang, Wei Tan, Xiaoming Xing, Jidong Lang. DL-CNV: A deep learning method for identifying copy number variations based on next generation target sequencing[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 202-215. doi: 10.3934/mbe.2020011







 DownLoad:
DownLoad: