Citation: Lan Zou, Jing Chen, Shigui Ruan. Modeling and analyzing the transmission dynamics of visceral leishmaniasis[J]. Mathematical Biosciences and Engineering, 2017, 14(5&6): 1585-1604. doi: 10.3934/mbe.2017082

| [1] | [ D. A. Ashford,J. R. David,M. Freire,R. David,I. Sherlock,M. D. C. Eulalio,D. P. Sampaio,R. Badaro, Studies on control of visceral leishmaniasis: Impact of dog control on canine and human visceral leishmaniasis in Jacobina, Bahia, Brazil, Am. J. Trop. Med. Hyg., 59 (1998): 53-57. |
| [2] | [ P. M. Boggiatto, K. N. Gibson-Corley and K. Metz, et. al., Transplacental transmission of Leishmania infantum as a means for continued disease incidence in North America, PLoS Negl. Trop. Dis. , 5 (2011), e1019. |
| [3] | [ P. M. Boggiatto,A. E. Ramer-Tait,K. Metz, Immunologic indicators of clinical progression during canine Leishmania infantum infection, Clin. Vaccine Immunol., 17 (2010): 267-273. |
| [4] | [ M. N. Burrattini,F. B. A. Cuoutinho,L. F. Lopez,E. Massad, Modeling the dynamics of leishmaniasis considering human, animal host and vector populations, J. Biol. Sys., 6 (1998): 337-356. |
| [5] | [ Chinese Center for Disease Control and Prevention, Public Health Data Center, 2004-2013, Available from: http://www.phsciencedata.cn/Share/index.jsp. |
| [6] | [ O. Courtenay, C. Carson, L. Calvo-Bado, L. M. Garcez and R. J. Quinnell, Heterogeneities in Leishmania infantum infection: using skin parasite burdens to identify highly infectious dogs, PLoS Negl. Trop. Dis., 8 (2014), e2583. |
| [7] | [ C. Dye, The logic of visceral leishmaniasis control, Am. J. Trop. Med. Hyg., 55 (1996): 125-130. |
| [8] | [ I. M. ELmojtaba,J. Y. T. Mugisha,M. H. A. Hashim, Mathematical analysis of the dynamics of visceral leishmaniasis in the Sudan, Appl. Math. Comput., 217 (2010): 2567-2578. |
| [9] | [ K. J. Esch,N. N. Pontes,P. Arruda,A. O'Connor,L. Morais,S. M. Jeronimo,C. A. Petersen, Preventing zoonotic canine leishmaniasis in northeastern Brazil: Pet attachment and adoption of community leishmania prevention, Am. J. Trop. Med. Hyg., 87 (2012): 822-831. |
| [10] | [ L. Gradoni, Canine leishmania vaccines: Still a long way to go, Vet. Parasitol., 208 (2015): 94-100. |
| [11] | [ T. Grinnage-Pulley, B. Scott and C. A. Petersen, A mother's gift: Congenital transmission of Trypanosoma and Leishmania species, PLoS Pathog., 12 (2016), e1005302. |
| [12] | [ N. Hartemink, S. O. Vanwambeke, H. Heesterbeek, D. Rogers, D. Morley, B. Pesson, C. Davies, S. Mahamdallie and P. Ready, Integrated mapping of establishment risk for emerging vector-borne infections: A case study of canine leishmaniasis in southwest France, PLoS One, 6 (2011), e20817. |
| [13] | [ G. Hasibeder,C. Dye,J. Carpenter, Mathematical modeling and theory for estimating the basic reproduction number of canine leishmaniasis, Parasitol., 105 (1992): 43-53. |
| [14] | [ Imperial College London, Theoretical Immunology Group Resources, Sand fly fact sheet, Available from: http://wwwf.imperial.ac.uk/theoreticalimmunology/exhibit2010/pdf/fs-sandflies.pdf. |
| [15] | [ S. F. Kerr,W. E. Grant,N. O. Dronen Jr, A simulation model of the infection cycle of Leishmania mexicana in Neotoma microbus, Ecol. Model., 98 (1997): 187-197. |
| [16] | [ Länger, et. al., Modeling of leishmaniasis infection dynamics: Novel application to the design of effective therapies, BMC Syst. Biol., 6 (2012), 1. |
| [17] | [ I. D. Lima,J. W. Queiroz,H. G. Lacerda, Leishmania infantum chagasi in northeastern Brazil: Asymptomatic infection at the urban perimeter, Am. J. Trop. Med. Hyg., 86 (2012): 99-107. |
| [18] | [ L. V. R. Lima,L. A. Carneiro,M. B. Campos, Canine visceral leishmaniasis due to Leishmania (L.) infantum chagasi in Amazonian Brazil: comparison of the parasite density from the skin, lymph node and visceral tissues between symptomatic and asymptomatic, seropositive dogs, Revista Institut. Med. Trop. Sao Paulo, 52 (2010): 259-265. |
| [19] | [ G. Michel,C. Pomares,B. Ferrua,P. Marty, Importance of worldwide asymptomatic carriers of leishmania infantum (L. chagasi) in human, Acta Trop., 119 (2011): 69-75. |
| [20] | [ R. Molina,J. M. Lohse,F. Pulido, Infection of sandflies by humans co-infected with leishmania infantum and human immuneodeficiency virus, Amer. J. Trop. Med. Hyg., 60 (1999): 51-53. |
| [21] | [ T. J. Naucke,S. Lorentz, Non-sandfly transmission of canine leishmaniasis, Tieraerztliche Umschau, 68 (2013): 121-125. |
| [22] | [ C. B. Palatnik-de-Sousa,I. Silva-Antunes,A. Morgado,I. Menz,M. Palatnik,C. Lavor, Decrease of the incidence of human and canine visceral leishmaniasis after dog vaccination with leishmune in Brazilian endemic areas, Vaccine, 27 (2009): 3505-3512. |
| [23] | [ R. Reithinger,P. G. Coleman,B. Alexander, Are insecticide-impregnated dog collars a feasible alternative to dog culling as a atrategy for controlling canine visceral leishmaniasis in Brazil?, Int. J. Parasitol., 34 (2004): 55-62. |
| [24] | [ G. A. Romero and M. Boelaert, Control of visceral leishmaniasis in Latin America -a systematic revies, PLoS Negl. Trop. Dis., 4 (2010), e584. |
| [25] | [ A. Stauch, R. R. Sakar and A. Picado, et. al., Visceral leishmaniasis in the Indian subcontinent: Modelling epidemiology and control, PLoS Negl. Trop. Dis., 5 (2011), e1405. |
| [26] | [ P. van den Driessche,J. Watmough, Reproduction numbers and sub-threshold endemic equilibria for compartmental models of disease transmission, Math. Biosci., 180 (2002): 29-48. |
| [27] | [ J. Wang, Y. Ha and C. Gao, et al., The prevalence of canine Leishmania infantum infection in western China detected by PCR and serological tests, Parasit. Vectors, 4 (2011), 69. |
| [28] | [ J. Wang,C. Gao,Y. Yang, An outbreak of the desert sub-type of zoonotic visceral leishmaniasis in Jiashi, Xinjiang Uygur Autonomous Region, People's Republic of China, Parasitol. Int., 59 (2010): 331-337. |
| [29] | [ The World Bank Group, Population, total, 2015, Available from: http://data.worldbank.org/indicator/SP.POP.TOTL?locations=BR. |
| [30] | [ World Health Organization, Number of cases of visceral leishmaniasis reported data by country, Available from: http://apps.who.int/gho/data/node.main.NTDLEISHVNUM?lang=en. |
| [31] | [ World Health Organization, Available from: http://www.who.int/leishmaniasis/resources/BRAZIL.pdf. |
| [32] | [ World Health Organization, World Health Organization, Leishmaniasis in high-burden countries: An epidemiological update based on data reported in 2014, Weekly Epid. Record, 91 (2016): 287-296. |
| [33] | [ S. Zhao,Y. Kuang,C. Wu,D. Ben-Arieh,M. Ramalho-Ortigao,K. Bi, Zoonotic visceral leishmaniasis transmission: Modeling, backward bifurcation, and optimal control, J. Math. Biol., 73 (2016): 1525-1560. |
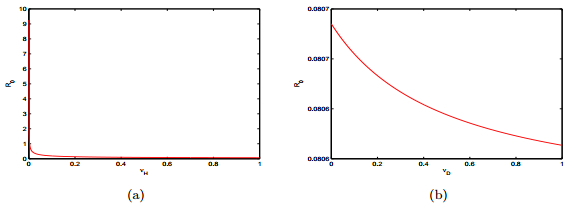
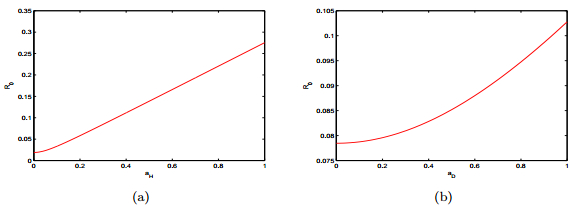

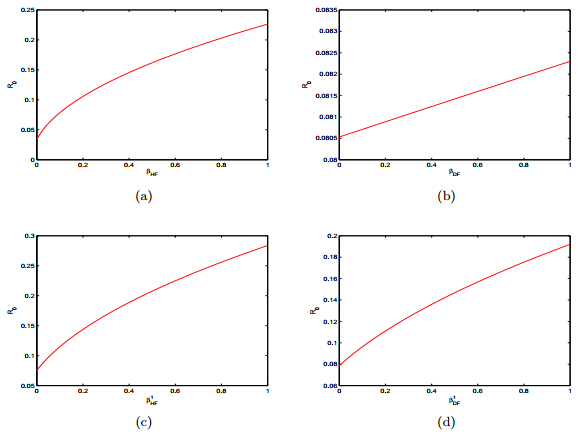
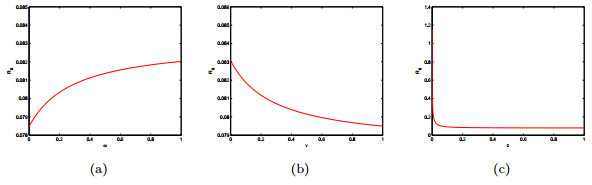


Figures(15) / Tables(2)

Lan Zou, Jing Chen, Shigui Ruan. Modeling and analyzing the transmission dynamics of visceral leishmaniasis[J]. Mathematical Biosciences and Engineering, 2017, 14(5&6): 1585-1604. doi: 10.3934/mbe.2017082







 DownLoad:
DownLoad: