Citation: Raghvendra P Singh, Guillaume Brysbaert, Marc F Lensink, Fabrizio Cleri, Ralf Blossey. Kinetic proofreading of chromatin remodeling: from gene activation to gene repression and back[J]. AIMS Biophysics, 2015, 2(4): 398-411. doi: 10.3934/biophy.2015.4.398

| [1] |
Jacob F, Monod J (1961) Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol 3: 318-356. doi: 10.1016/S0022-2836(61)80072-7

|
| [2] | Ptashne M (2004) A Genetic Switch: Phage Lambda Revisited (3rd. ed.), Cold Spring Harbor Laboratory Press, USA. |
| [3] |
Ackers GK, Johnson AD, Shea MA (1982) Quantitative model for gene regulation by lambda phage repressor. Proc Natl Acad Sci (USA) 79: 1129-1133. doi: 10.1073/pnas.79.4.1129
|
| [4] |
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41-45. doi: 10.1038/47412
|
| [5] | Narlikar GJ, Sundaramoorthy R, Owen-Hughes T (2013) Mechanisms and Functions of ATP Dependent Chromatin-Remodeling Enzymes. Cell 154: 440-503. |
| [6] |
Blossey R, Schiessel H (2008) Kinetic proofreading of gene activation by chromatin remodeling. HFSP Journal 2: 167-170. doi: 10.2976/1.2909080
|
| [7] |
Narlikar GJ (2010) A proposal for kinetic proof reading by ISWI family chromatin remodeling motors. Curr Opin Chem Biol 14: 660-665. doi: 10.1016/j.cbpa.2010.08.001
|
| [8] |
Blossey R, Schiessel (2011) Kinetic Proofreading in Chromatin Remodeling: The Case of ISWI/ACF. Biophys J 101: L30-L32. doi: 10.1016/j.bpj.2011.07.001
|
| [9] |
Herschlag D, Johnson FB (1993) Synergism in Transcriptional Activation: A Kinetic View. Genes Development 7: 173-179. doi: 10.1101/gad.7.2.173
|
| [10] |
Cosgrove MS, Boeke JD, Wolberger C (2004) Regulated Nucleosome Mobility and the Histone Code. Nature Struct Mol Biol 11: 1037-1043. doi: 10.1038/nsmb851
|
| [11] | Rippe K, Schrader A, Riede P, Strohner R, Lehmann E, Langst G (2007) DNA sequenceand conformation-directed positioning of nucleosomes by chromatin-remodeling complexes Proc Natl Acad Sci (USA) 104: 15635-15640. |
| [12] |
Erdel F, Schubert T, Marth C, Langst G, Rippe K (2010) Human ISWI chromatinremodeling complexes sample nucleosomes via transient binding reactions and become immobilized at active sites. Proc Natl Acad Sci (USA) 107: 19873-19878. doi: 10.1073/pnas.1003438107
|
| [13] |
Michel D (2011) Hierarchical cooperativity mediated by chromatin remodeling; the model of the MMTV transcription regulation. J Theoret Biol 287: 74-81. doi: 10.1016/j.jtbi.2011.07.020
|
| [14] |
Hopfield JJ (1974) Kinetic Proofreading: A New Mechanism for Reducing Errors in Biosynthetic Processes Requiring High Specificity. Proc Natl Acad Sci (USA) 71: 4135-4139. doi: 10.1073/pnas.71.10.4135
|
| [15] |
Ninio J (1975) Kinetic amplification of enzyme discrimination. Biochimie 57: 587-595. doi: 10.1016/S0300-9084(75)80139-8
|
| [16] |
Ferreira H, Flaus A, Owen-Hughes T (2007) Histone modifications in uence the action of Snf2 family remodelling enzymes by different mechanisms. J Mol Biol 374: 563-579. doi: 10.1016/j.jmb.2007.09.059
|
| [17] |
Agalioti T, Lomvardes S, Parekh B, Yie J, Maniatis T, Thanos D (2000) Ordered Recruitment of Chromatin Modifying and Basal Factors to the IFN-b promoter. Cell 103: 667-678. doi: 10.1016/S0092-8674(00)00169-0
|
| [18] |
Lomvardas S, Thanos D (2001) Nucleosome sliding via TBP DNA binding in vivo. Cell 106: 685-696. doi: 10.1016/S0092-8674(01)00490-1
|
| [19] |
Agalioti T, Chen G, Thanos D (2002) Deciphering the Transcriptional histone acetylation code for a human gene. Cell 111: 381-392. doi: 10.1016/S0092-8674(02)01077-2
|
| [20] |
Koutroubas G, Merika M, Thanos D (2008) Bypassing the requirements for epigenetic modi fications in gene transcription by increasing enhancer strength. Mol Cell Biol 28: 926-938. doi: 10.1128/MCB.01344-07
|
| [21] |
Nolis IK, McKay DJ, Mantouvalou E, Lomvardas S, Merika M, Thanos D (2009) Transcription factors mediate long range enhancer-promoter interactions. Proc Natl Acad Sci (USA) 106: 20222-20227. doi: 10.1073/pnas.0902454106
|
| [22] |
Ford E, Thanos D (2010) The transcriptional code of human IFN-beta gene expression. Biochim Biophys Acta 1799: 328-336. doi: 10.1016/j.bbagrm.2010.01.010
|
| [23] | Alon U (2006) An Introduction to Systems Biology: Design Principles of Biological Circuits, Chapman & Hall/CRC, United Kingdom. |
| [24] |
Clapier CR, Cairns BR (2009) The biology of chromatin remodeling complexes. Annu Rev Biochem 78: 273-304. doi: 10.1146/annurev.biochem.77.062706.153223
|
| [25] |
Clapier CR, Cairns BR (2012) Regulation of ISWI involves inhibitory modules antagonized by nucleosomal epitopes. Nature 492: 280-285. doi: 10.1038/nature11625
|
| [26] |
Yang JG, Madrid TS, Sevastopoulos E, Narlikar GJ (2006) The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat Struct Mol Biol 13: 1078-1083. doi: 10.1038/nsmb1170
|
| [27] |
Racki LR, Yang JG, Nariman N, Partensky PD, Acevedo A, Purcell TJ, Cooke R, Cheng Y, Narlikar GJ (2009) The chromatin remodeller ACF acts as a dimeric motor to space nucleosomes. Nature 462: 1016-1021. doi: 10.1038/nature08621
|
| [28] |
Blosser TR, Yang JG ,Stone MD, Narlikar GJ, Zhuang X (2009) Dynamics of nucleosome remodeling by individual ACF complexes. Nature 462: 1022-1028. doi: 10.1038/nature08627
|
| [29] |
Brysbaert G, Lensink MF, Blossey R (2015) Regulatory motifs on ISWI chromatin remodelers: molecular mechanisms and kinetic proofreading. J Phys Condens Matter 27: 064108. doi: 10.1088/0953-8984/27/6/064108
|
| [30] | Teif VB, Kepper N, Yserentant K, Wiedemann G, Rippe K (2015) Affinity, stoichiometry and cooperativity of heterochromatin protein 1 (HP1) binding to nucleosomal arrays, J Phys Condens Matter 27: 064110. |
| [31] |
Lesne A, Foray N, Cathala G, Fornée T, Wong H, Victor JM (2015) Chromatin fiber allostery and the epigenetic code. J Phys Condens Matter 27: 064114. doi: 10.1088/0953-8984/27/6/064114
|
| [32] |
Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Mller S, Pawson T, Gingras AC, Arrowsmith CH, Knapp S. (2012) Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149: 214-231. doi: 10.1016/j.cell.2012.02.013
|
| [33] |
Josling G, Selvarajah SA, Petter M, Duffy MF (2012) The Role of Bromodomain Proteins in Regulating Gene Expression. Genes 3: 320-343. doi: 10.3390/genes3020320
|
| [34] |
Owen DJ, Ornaghi P, Yang JC, Lowe N, Evans PR, Ballario P, Neuhaus D, Filetici P, Travers AA (2000) The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase Gcn5p. EMBO J 19: 6141-6149. doi: 10.1093/emboj/19.22.6141
|
| [35] |
Hudson BP, Martinez-Yamout MA, Dyson HJ, Wright PE (2000) Solution structure and acetyl-lysine binding activity of the GCN5 bromodomain. J Mol Biol 304: 355-370. doi: 10.1006/jmbi.2000.4207
|
| [36] | Singh RP (2013) Computer Modeling of the Application of Mechanical Forces to Biomolecules. Thesis University of Lille. |
| [37] |
Ponder JW, Richards FM (1987) An efficient Newton-like method for molecular mechanics energy minimization of large molecules. J Comput Chemistry 8: 1016-1024. doi: 10.1002/jcc.540080710
|
| [38] |
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 25: 1605-1612. doi: 10.1002/jcc.20084
|
| [39] |
Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, exible, and free. J Comput Chem 26: 1701-1718. doi: 10.1002/jcc.20291
|
| [40] | Hess HB, Berendsen HJC, Fraaije JGEM (1997) LINCS: A linear constraint solver for molecular simulations. J Comp Chem 18: 1463-1472. |
| [41] |
Miyamoto S, Kollman PA (1992) Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem 13: 952-962. doi: 10.1002/jcc.540130805
|
| [42] | Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ AutoDock4 and AutoDockTools4: Automated docking with selective receptor exibility. J Comput Chem 30: 2785-2761. |
| [43] | Trott O, Olson AJ (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading J Comput Chem 31: 455-461. |
| [44] |
Isralewitz B, Baudry J, Gullingsrud J, Kosztin D, Schulten K (2001) Steered molecular dynamics investigations of protein function. J Mol Graph Model 19: 13-25. doi: 10.1016/S1093-3263(00)00133-9
|
| [45] |
Isralewitz B, Gao M, Schulten K (2001) Steered molecular dynamics and mechanical functions of proteins. Curr Opin Struct Biol 11: 224-230. doi: 10.1016/S0959-440X(00)00194-9
|
| [46] |
Torrie GM, Valleau JP (1977) Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J Comp Phys 23: 187-199. doi: 10.1016/0021-9991(77)90121-8
|
| [47] |
Hummer G, Szabo A (2010) Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc Natl Acad Sci (USA) 107: 21441-21446. doi: 10.1073/pnas.1015661107
|
| [48] |
Cieniewicz AM, Moreland L, Ringel AE, Mackintosh SG, Raman A, Gilbert TM, Wolberger C, Tackett AJ, Taverna SD (2014) The bromodomain of Gcn5 regulates site specificity of lysine acetylation on histone H3. Mol Cell Proteomics 13: 2896-2910. doi: 10.1074/mcp.M114.038174
|
| [49] |
Jiang J, Lu J, Lu D, Liang Z, Li L, Ouyang S, Kong X, Jiang H, Shen B, Luo C (2012) Investigation of the Acetylation Mechanism by GCN5 Histone Acetyltransferase. PloS One 7: e36660. doi: 10.1371/journal.pone.0036660
|
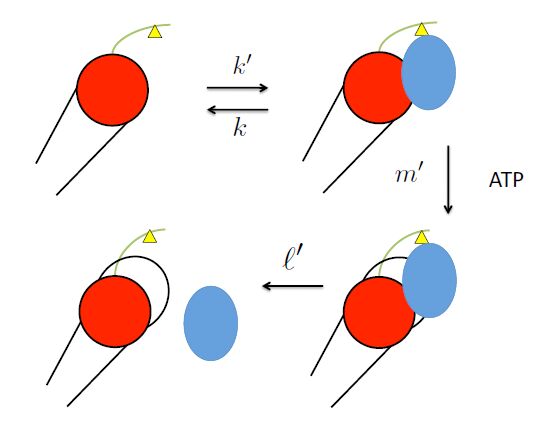
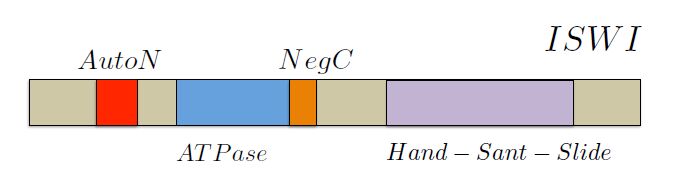
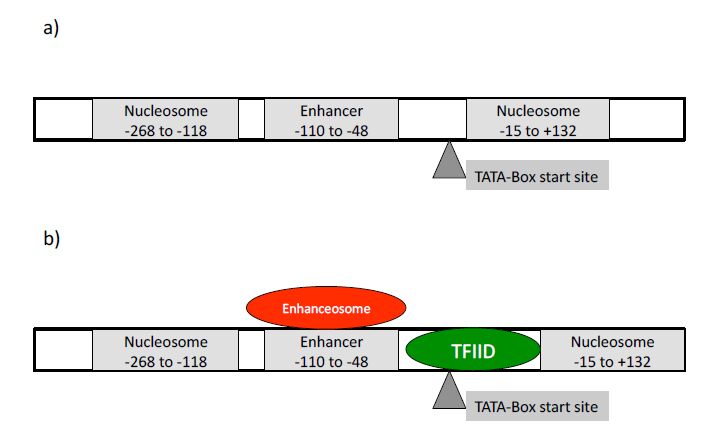
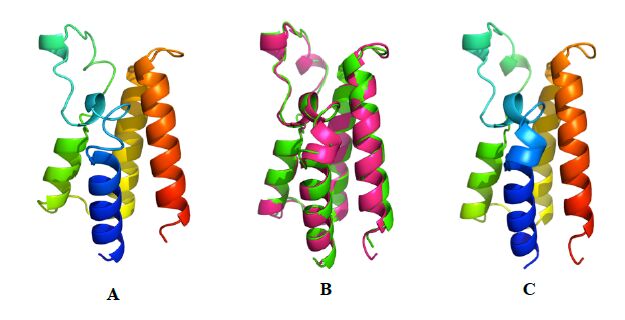
Figures(5) / Tables(1)

Raghvendra P Singh, Guillaume Brysbaert, Marc F Lensink, Fabrizio Cleri, Ralf Blossey. Kinetic proofreading of chromatin remodeling: from gene activation to gene repression and back[J]. AIMS Biophysics, 2015, 2(4): 398-411. doi: 10.3934/biophy.2015.4.398







 DownLoad:
DownLoad: