Citation: Davide Sala, Andrea Giachetti, Antonio Rosato. Molecular dynamics simulations of metalloproteins: A folding study of rubredoxin from Pyrococcus furiosus[J]. AIMS Biophysics, 2018, 5(1): 77-96. doi: 10.3934/biophy.2018.1.77
| [1] | Mathieu F. M. Cellier . Evolutionary analysis of Slc11 mechanism of proton-coupled metal-ion transmembrane import. AIMS Biophysics, 2016, 3(2): 286-318. doi: 10.3934/biophy.2016.2.286 |
| [2] | Ken Takahashi, Takayuki Oda, Keiji Naruse . Coarse-grained molecular dynamics simulations of biomolecules. AIMS Biophysics, 2014, 1(1): 1-15. doi: 10.3934/biophy.2014.1.1 |
| [3] | Stephanie H. DeLuca, Samuel L. DeLuca, Andrew Leaver-Fay, Jens Meiler . RosettaTMH: a method for membrane protein structure elucidation combining EPR distance restraints with assembly of transmembrane helices. AIMS Biophysics, 2016, 3(1): 1-26. doi: 10.3934/biophy.2016.1.1 |
| [4] | Arjun Acharya, Madan Khanal, Rajesh Maharjan, Kalpana Gyawali, Bhoj Raj Luitel, Rameshwar Adhikari, Deependra Das Mulmi, Tika Ram Lamichhane, Hari Prasad Lamichhane . Quantum chemical calculations on calcium oxalate and dolichin A and their binding efficacy to lactoferrin: An in silico study using DFT, molecular docking, and molecular dynamics simulations. AIMS Biophysics, 2024, 11(2): 142-165. doi: 10.3934/biophy.2024010 |
| [5] | Sebastian Kube, Petra Wendler . Structural comparison of contractile nanomachines. AIMS Biophysics, 2015, 2(2): 88-115. doi: 10.3934/biophy.2015.2.88 |
| [6] | Tika Ram Lamichhane, Hari Prasad Lamichhane . Structural changes in thyroid hormone receptor-beta by T3 binding and L330S mutational interactions. AIMS Biophysics, 2020, 7(1): 27-40. doi: 10.3934/biophy.2020003 |
| [7] | Ashwani Kumar Vashishtha, William H. Konigsberg . The effect of different divalent cations on the kinetics and fidelity of Bacillus stearothermophilus DNA polymerase. AIMS Biophysics, 2018, 5(2): 125-143. doi: 10.3934/biophy.2018.2.125 |
| [8] | Juliet Lee . Insights into cell motility provided by the iterative use of mathematical modeling and experimentation. AIMS Biophysics, 2018, 5(2): 97-124. doi: 10.3934/biophy.2018.2.97 |
| [9] | Chia-Wen Wang, Meng-Han Lin, Wolfgang B. Fischer . Cholesterol affected dynamics of lipids in tailor-made vesicles by ArcVes software during multi micro second coarse grained molecular dynamics simulations. AIMS Biophysics, 2023, 10(4): 482-502. doi: 10.3934/biophy.2023027 |
| [10] | Yohei Matsunaga, Tsuyoshi Kawano . The C. elegans insulin-like peptides (ILPs). AIMS Biophysics, 2018, 5(4): 217-230. doi: 10.3934/biophy.2018.4.217 |
Constant advances in computational power and methods have opened the possibility to study many biological processes using in silico methods. In particular, the massive computing efficiency of modern GPGPUs can be exploited in molecular dynamics (MD) simulations to explore at the atomistic level timescales of motion that are relevant to functional properties [1,2]. One of the processes that in principle can be studied in a cost-effective way using MD is the folding mechanism [3]. Protein folding occurs when an unstructured polypeptide chain reaches its stable and functional three-dimensional structure. For different proteins, this can happen in a broad range of timescales from microseconds to seconds and higher. Thus, to obtain sufficient sampling to meaningfully comment on folding mechanisms by MD simulations remains a challenging task, notwithstanding the recent advances in computing power, MD methods and accuracy of force-fields [4]. In fact, the simulation length must be at least on the microsecond timescale to stand a good chance of observing a single folding event [5,6]. Achieving enough folding/unfolding transitions to define the folding pathway and accurately measure kinetic and thermodynamic quantities is even more demanding [7]. Alternatively, MD simulations for the folded and unfolded states can be carried out independently [8]. Despite the common difficulties mentioned above, the discovery of fast-folding proteins has provided systems for which the achievable MD timescales match the experimental folding times [9,10]. As a result, the combination of experimental and theoretical studies contributed significantly to our knowledge of the thermodynamics and kinetics of folding [11].
Despite the importance of metalloproteins in many important biological functions [12], little attention has been devoted to the theoretical investigation of metal-induced folding. This is mainly due to the difficulties to correctly model metal-protein interactions [13]. One of the unsolved questions regards how the presence of the metal affects the protein folding in terms of structural and dynamics properties [14]. In this work, we analyzed the folding mechanism of a highly stable metalloprotein: rubredoxin from Pyrococcus furiosus (PfRd). PfRd is a globular protein of 53 amino acids that binds a single iron ion in the +2 or +3 oxidation state [15]. The iron atom is coordinated to the protein through cysteinyl sulfurs of two consensus cysteine motifs, CXXC and CPXC. Various studies have addressed the structural basis of PfRd hyperthermostability as well as its ability to refold both in presence and absence of iron [16,17,18,19]. The secondary and tertiary structure of PfRd devoid of its iron cofactor (apo-PfRd) are very similar to iron-loaded PfRd (holo-PfRd) [20]. However, experimental studies of Holo- and apo-PfRd showed significant different in their unfolding and refolding processes. Apo-PfRd reaches about 50% unfolding at about 343 K, with the folded 50% of the molecules still displaying structural features consistent with the native structure of the holo-protein. The unfolding process is reversible [20]. On the other hand, the unfolding of holo-PfRd is essentially irreversible [21]. This has been ascribed to presence of the iron binding site; a designed variant of the protein that retains the tertiary structure and thermostability of wild-type PfRd but cannot bind iron features reversible folding [22]. Addition of iron to apo-PfRd in the presence of denaturing agents such as either 6 M urea or 6 M guanidine hydrochloride triggers protein refolding eventually yielding correctly folded holo-PfRd [19]. However, this does not happen if the denaturing agents are removed prior to addition of the metal. More recently, it has been suggested that these subtleties in the refolding process of apo- and holo-PfRd depend on the extent of structure loss upon removal of the iron ion from the holo-protein, which in turn depends on the amount of denaturant added. This is crucial to get the correct packing of the hydrophobic core residues during protein refolding [16].
In this work, we aimed at evaluating the folding properties of holo-PfRd. We applied two different MD methods: a classical MD (cMD) simulation in explicit solvent and an accelerated MD (aMD) simulation where a biased potential enhances the conformational sampling [23,24]. A well-known problem in applying cMD for folding simulations is that proteins usually have high barriers that separate the minima of the potential energy surface. Consequently, the system is often trapped in a local minimum for long periods of simulation time, preventing extensive exploration of the potential energy surface. Eventually, this may prevent reaching the native folded state of the protein starting far away from the global minimum. Even achieving a timescale comparable with the experimental data is not guarantee of success. To overcome these limitations, in this work we applied the aMD method, by which the height of local barriers is reduced, thus allowing the calculation to evolve much faster [25].
This work is based on MD simulations performed using the Amber package of molecular simulation programs [26]. All the MD runs were prepared from the crystal structure of holo-PfRd (PBD 1BRF). The apo-form of the protein was obtained by removing the iron ion from the metal binding site. The holo-form has an iron ion covalently bound to 4 cysteine side chains. Thus, the RESP charges compatible with the AMBER force field for the metal center were used [27]. The systems were solvated with TIP3P water model molecules and the overall charge balanced. The forcefield used was Amber14SB. The starting structure of the folding simulations is derived from a brief MD at 600 K. More precisely, the most elongated structure was chosen as representative of the unfolded state. The MD runs were prepared following the same basic protocol consisting in 4 steps: water minimization, system heating to 300 K in NVT and density equilibration in NPT conditions. All the production runs were performed on a Nvidia Tesla K20m GPGPU applying a PME cutoff of 10 Å. This computational infrastructure is available to users via the AMPS-NMR portal within the West-Life (www.west-life.eu) project [28,29].
The first production run was performed on the folded state of the apo-form for 1 µs (APO). Then, a classical MD run was carried out for 1 µs to fold the protein (F-cMD). In addition, to increase the probability of sampling a folding event the accelerated molecular dynamics method was applied (F-aMD). Accelerated molecular dynamics was carried out starting from the same unfolded structure used for the cMD run. Differently form cMD, during the aMD run the whole potential is boosted as follows:
| ΔVP= (EP−(ΔVD))2αP+(EP−(V+ΔVD)) | (1) |
where the torsion potential ΔVD is given by
| ΔVD=(ED−V)2αD+(ED−V) | (2) |
The terms EP and ED define the average potential and dihedral energies. The terms αP and αD are the inverse strength boost factors for the total and dihedral potential energy, respectively. Prior to the production run a classical MD was carried out for 50 ns to define reasonable constants. As a result, the following values were applied: EP = −47.64 kcal/mol, ED = 814 kcal/mol, αP = 2.542 and αD = 42. The production run was performed for 11.6 µs.
The Root Mean Square Fluctuation (RMSF) is an index to measure the structural flexibility. It is defined by
| RMSF=√∑τt=1∥r(t)−〈r〉∥2τ | (3) |
where τ is the number of frames, r(t) is the atomic position at time t and 〈r〉 is the average structure. Basically, this parameter corresponds to the standard deviation of the atomic positions from the average structure over a trajectory.
The order parameter (S2) measures the magnitude of the angular fluctuation of a chemical bond vector such as the NH bond, thus reflecting the flexibility of a protein specific site. We used two different methods to calculate the order parameter of NH vectors. The isotropic reorientational eigenmode dynamics (iRED) method relies on a principal component analysis of the isotropically averaged covariance matrix [28]. In the second method, the autocorrelation function (ACF) of the vectors is fitted on a monoexponential curve [29].
The content of secondary structure along the trajectories was measured using the DSSP program [30,31]. Its dictionary consists of 8 classes: random-coil, 3-turn helix, α helix, 5-turn helix, turn, beta-sheet, beta-bridge and bend. In this work, the three helical topologies are indicated with “helix” and the beta-structures as “β-sheet”.
The Root Mean Square Deviation (RMSD) is a conformational distance index. It is defined by
| RMSD=√∑Ni=1mi∥r(a)i−r(b)i∥2∑Ni=1mi | (4) |
where N are the atoms selected, ri is the position vector and a and b are two different conformations. In this study, all RMSD values were referred to the crystal structure.
Chemical shifts were predicted using the PPM chemical shifts prediction web server (http://spin.ccic.ohio-state.edu) that was parametrized specifically for MD trajectories [32].
The Solvent Accessibility Surface Area (SASA) is a measure of the exposure to the solvent. For the common water, a probe of 1.4 Å is used to scan the molecular surface. In this study, the SASA is calculated on selected hydrophobic residues belonging to the protein core (residues Trp3, Tyr10, Tyr12, Phe29, Trp36, Phe48, Ile23 and Leu32).
For the present work, we defined “native contact” any contact shorter than a defined cut-off that is present in the reference structure. Only the contacts among core hydrophobic residues closer than 7 Å were considered.
The Principal Component Analysis was performed on the Cα atoms of the F-aMD and APO trajectories. The conformations were fitted on the crystal structure and separated on the average structure. The first two eigenvectors include 44% of the motions.
PfRd has a globular shape, with the iron ion covalently bound to 4 cysteine side chains (residues 5, 8, 38 and 41) and is known to be quite rigid in its folded holo form, as shown by the low values of the B factor [33]. Its conserved secondary structure consists of three 310-helices (residues 19-21, 29-31 and 45-47) and one antiparallel triple stranded β-sheet (residues 2-5, 11-13 and 48-50). The hydrophobic core consists of 8 residues (residues Trp3, Tyr10, Tyr12, Phe29, Trp36, Phe48, Ile23 and Leu32). In this work, we attempted to simulate the folding process of holo-PfRd, starting from an extended structure. To this end, two different simulation schemes were adopted (Table 1). In addition, we simulated the dynamics of apo-PfRd; the starting model for the latter was the folded structure (PDB 1BRF) after removal of the metal ion.
| Type of simulation | Simulation length (µs) | Starting structure |
| Classical-Apo (APO) | 1 | Crystal structure without metal ion |
| Folding Accelerated (F-aMD) | 11.6 | Unfolded state with the metal bound |
| Folding Classical (F-cMD) | 1 | Unfolded state with the metal bound |
| The acronyms used in the text to identify each simulation are given in brackets in the first column. Both folding runs started from the same elongated conformation with the iron ion bound. | ||
 DownLoad: CSV
DownLoad: CSVThe Root Mean Square Fluctuation (RMSF) analysis of the backbone atoms provides a measure of the flexibility of residues with respect to the average structure (Figure 1A). The higher the RMSF of one residue, the higher its mobility. In addition, protein motions can be summarized by calculating the order parameter (S2) of N-H vectors [34,35]. Other bond vectors can also be used [36,37]. In the APO trajectory, we estimated the S2 values of N-H vectors by two different methods: the IRED analysis [28] and a more traditional approach where the autocorrelation function (ACF) is fitted to a monoexponential curve [29] (Figure 1B). Despite the absence of the metal coordination by Cys5 and Cys8, the first 8 residues of the N-terminus are very rigid whereas Gly9 has higher flexibility than nearby residues in the RMSF profile. Residues 11 to 13 are quite rigid, and then the RMSF increases and reaches the maximum value in the region 20-21, whereas the order parameters values are still close to the overall protein average. From 22 to 29 the RMSF profile decreases progressively, but then increases again from residue 30 until the peak of the region 34-35. Then, the flexibility drops rapidly for the subsequent two residues. Differently from the RMSF, the order parameter profiles in this central region of the protein have more variability suggesting that the local environment affects the dynamics of the N-H vectors more than larger-scale motions. The absence of the covalently bound iron ion affects in a similar way both the RMSF and the order parameters in the region limited by the metal binding Cys38 and Cys41. In fact, all the analyses revealed high mobility in this region. The RMSF and S2 profiles agree also in the last protein segment showing a modest flexibility from residue 42 to 51 followed by enhanced mobility for the last two residues of the C-terminus.
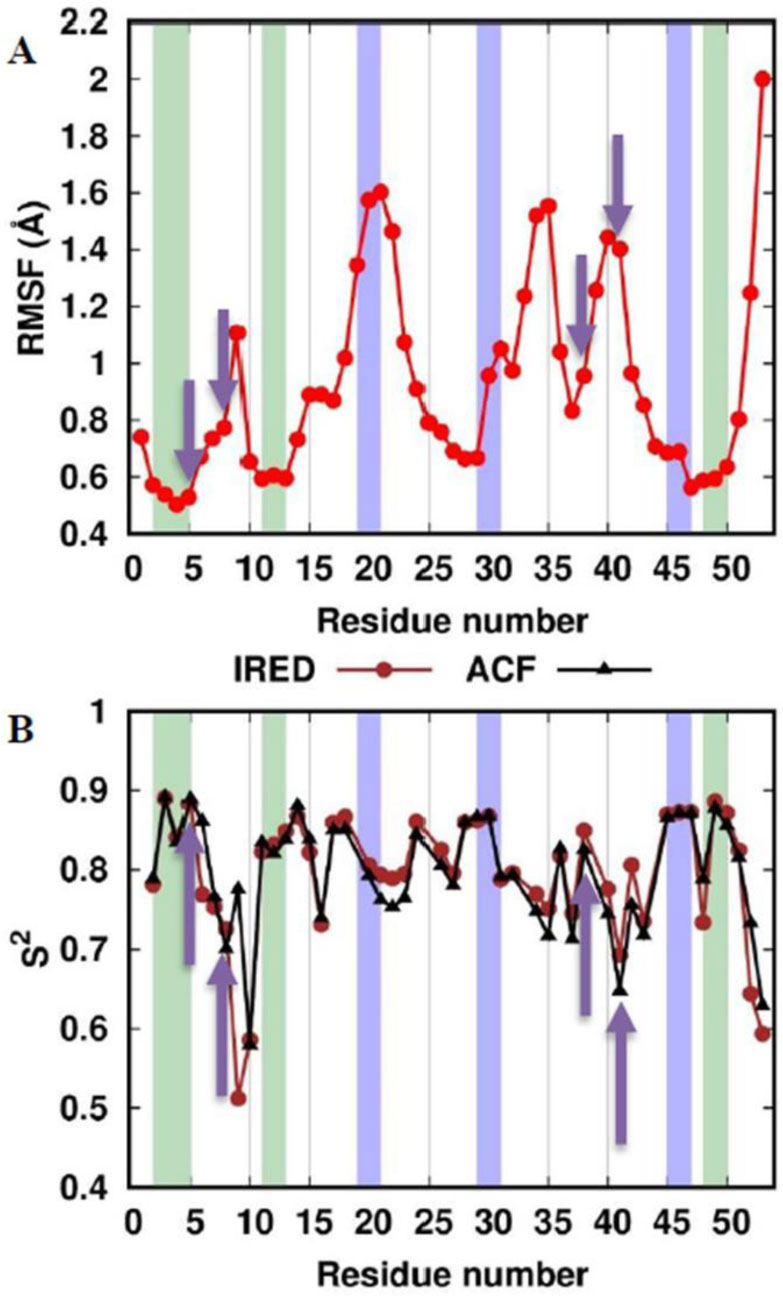 Figure 1. RMSF and order parameter profiles of the APO trajectory. The residues corresponding to β-sheets and 310-helices in the crystal structure are colored with a green and blue background, respectively. The metal-binding cysteines are indicated with yellow arrows. (A) RMSF of the backbone atoms. (B) The NH vector S2 profiles computed with the IRED method (IRED, brown) and by calculating the autocorrelation function (ACF, black).
Figure 1. RMSF and order parameter profiles of the APO trajectory. The residues corresponding to β-sheets and 310-helices in the crystal structure are colored with a green and blue background, respectively. The metal-binding cysteines are indicated with yellow arrows. (A) RMSF of the backbone atoms. (B) The NH vector S2 profiles computed with the IRED method (IRED, brown) and by calculating the autocorrelation function (ACF, black).The apo-PfRd structure maintained its native (i.e. present in the crystal) secondary structure elements for most of the simulation (Figure 2). The Root Mean Square Deviation (RMSD) from the crystal structure of the backbone atoms shows a great stability along the trajectory, with an average value lower than 2.5 Å and only a modest increase in the second half of the simulation (Figure 3). The three-stranded β-sheet is present in almost the totality of the frames. On the contrary, the three helices are not as conserved as the triple strand. In particular, the first helix is present in just 23% of the snapshots against 58% and 67% of the second and third helix, respectively. A more detailed analysis revealed that most of the helix-missing frames sampled a turn structure featuring hydrogen bonds typical of helices (
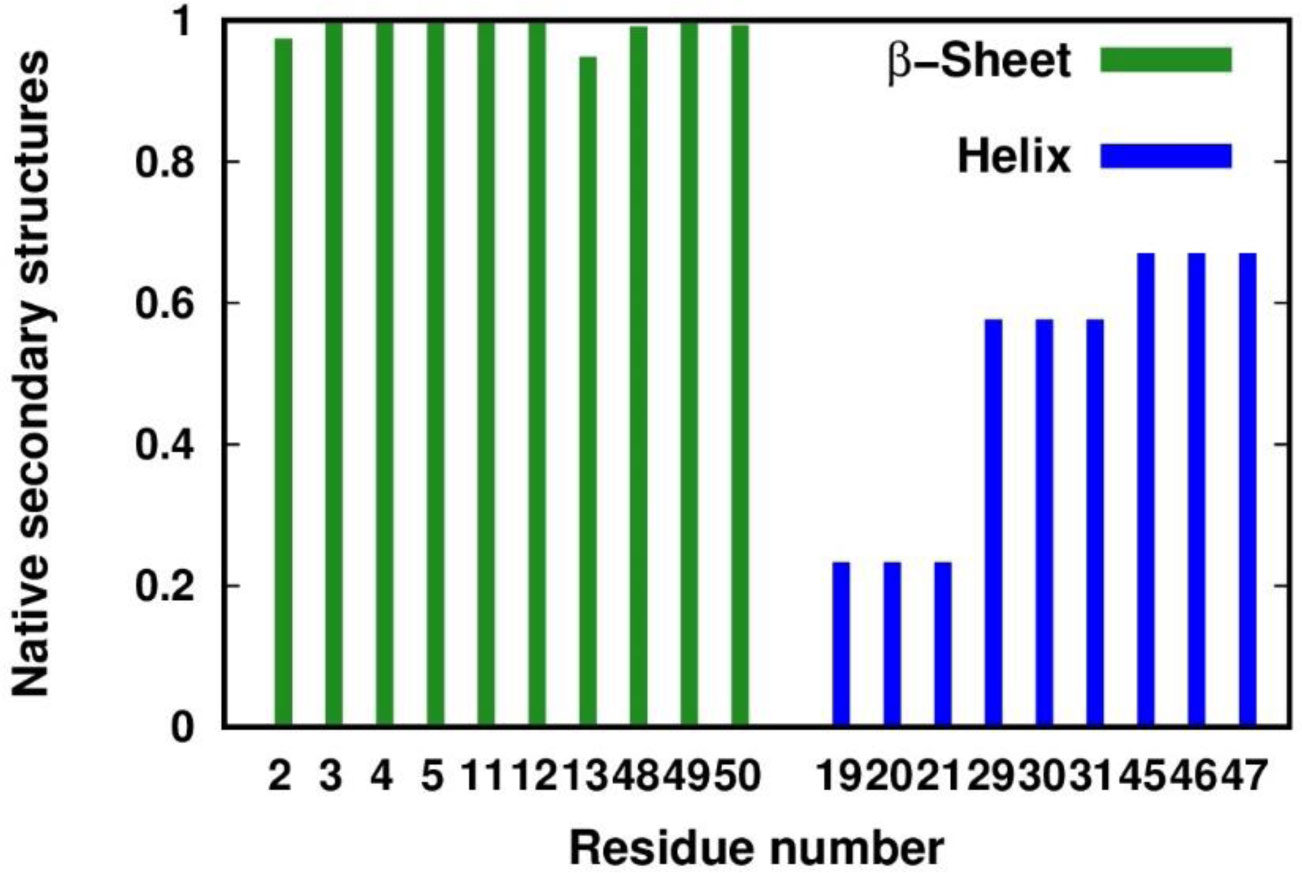 Figure 2. Fraction of native secondary structures sampled in the APO simulation. For each residue in β-sheet (green) or helix (blue) structure in the crystal structure, we report the fraction of conformations with the same secondary structure.
Figure 2. Fraction of native secondary structures sampled in the APO simulation. For each residue in β-sheet (green) or helix (blue) structure in the crystal structure, we report the fraction of conformations with the same secondary structure.To evaluate the deviation of the APO conformational ensemble from the behavior of the holo-form, we used the NMR data of the latter [40,41,42]. We compared the chemical shifts of the Cα, C and N atoms computed from the trajectory with the available experimental data [38]. Figure 4 reports the correlation of the predicted values and the experimental values. The Cα atoms display a very high correlation, with a value of the Pearson coefficient as high as 0.98. The N and C atoms have Pearson coefficients of 0.87 and 0.73, respectively. In general, the correlation is good for all the atoms assessed, confirming that the apo-form samples conformations that are consistent with the holo-form.
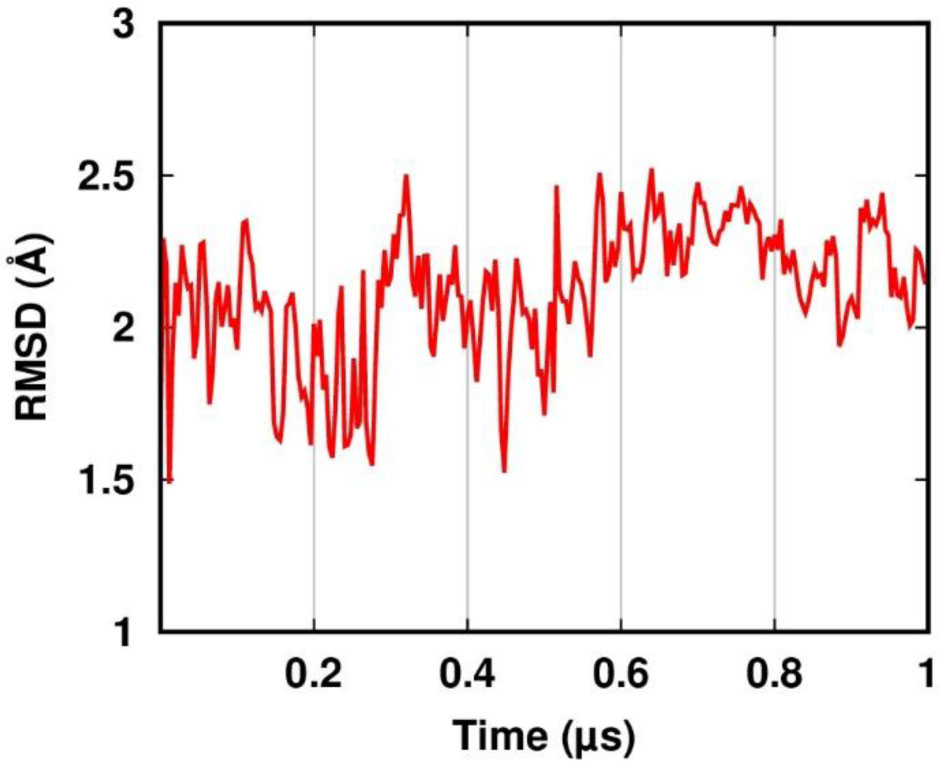 Figure 3. Root Mean Square Deviation from the crystal structure in the APO simulation.
Figure 3. Root Mean Square Deviation from the crystal structure in the APO simulation.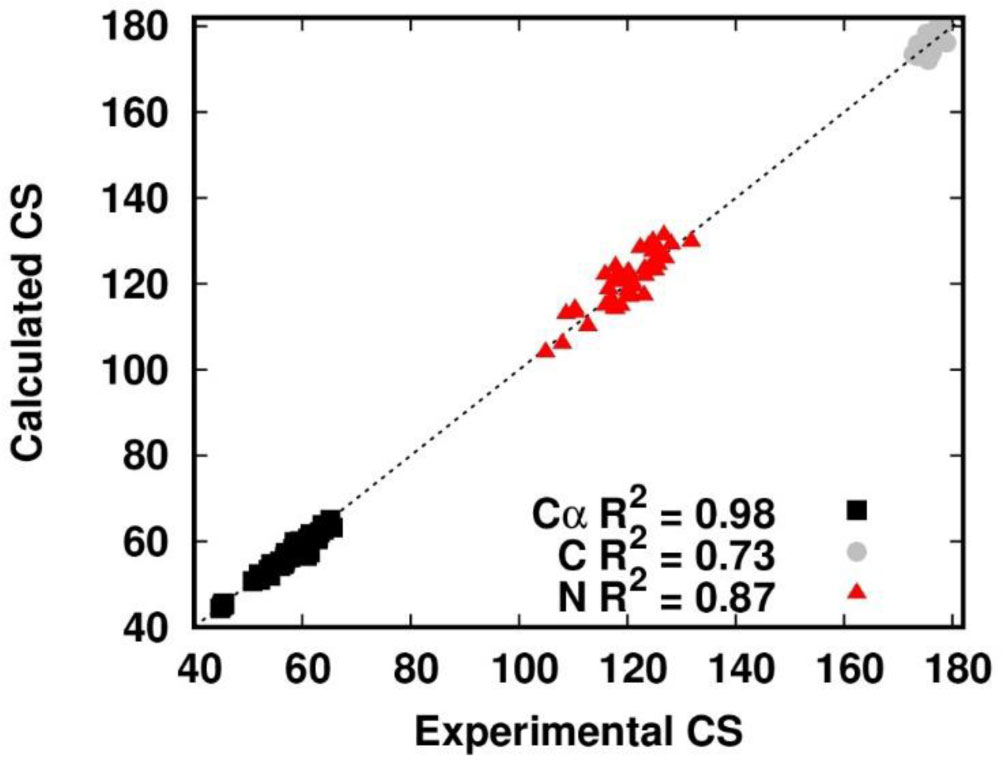 Figure 4. Correlation between experimental and calculated chemical shifts (CS) in the APO simulation. The Cα atoms are shown as black squares. The C atoms are shown as gray circles. The N atoms are shown as red triangles. The dashed line is y = x and is shown only to guide the eye.
Figure 4. Correlation between experimental and calculated chemical shifts (CS) in the APO simulation. The Cα atoms are shown as black squares. The C atoms are shown as gray circles. The N atoms are shown as red triangles. The dashed line is y = x and is shown only to guide the eye.In summary, apo-PfRd features a great structural stability. Nevertheless, the lack of metal binding affects the dynamics of local structural elements. For instance, the anomalous flexibility of Gly9 compared with its nearby residues could be the result of the destabilization of the aromatic sidechains in the protein core (Figure 5). Furthermore, some regions sampled non-native secondary structures that can result in a discrepancy between the S2 and RMSF profiles. This is the case of residues 19-22 and 29-31 (i.e. in correspondence of the first and second native helix) where the RMSF plot shows a relatively high flexibility not detected by the S2 analysis. The difference is related to the native helices being replaced by H-bonded turns (
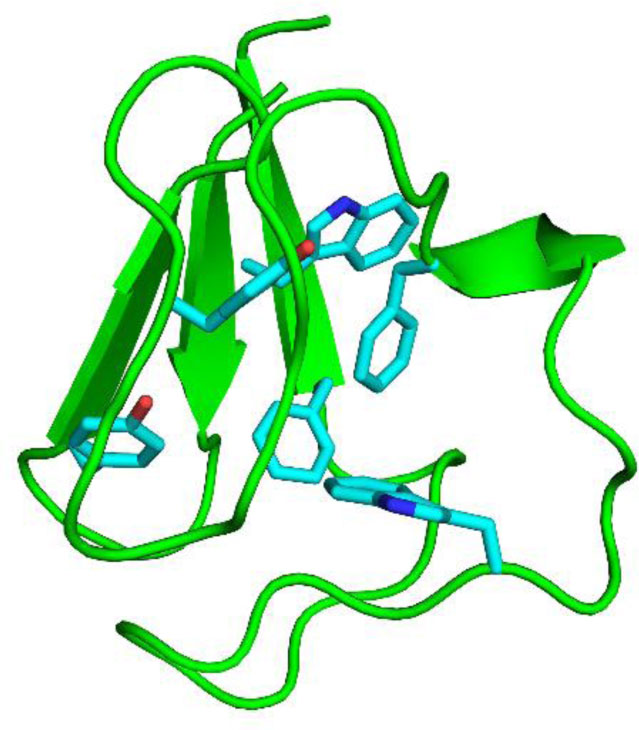 Figure 5. Snapshot at 1 µs showing the 3-D structure of the apo-PfRd. The aromatic side chains in the protein core are shown in cyan (Trp3, Tyr10, Tyr12, Phe29, Trp36 and Phe48).
Figure 5. Snapshot at 1 µs showing the 3-D structure of the apo-PfRd. The aromatic side chains in the protein core are shown in cyan (Trp3, Tyr10, Tyr12, Phe29, Trp36 and Phe48).The folding process of holo-PfRd was simulated using two different computational strategies: classical MD (F-cMD) and accelerated MD (F-aMD) (Table 1). The purpose of these simulations was to achieve a structure as close as possible to the folded state starting from an unfolded conformation, where we enforced metal coordination.
Figure 6 shows the S2 profile for the simulations. As expected, the boost applied in the F-aMD simulation makes the average S2 values for this simulation lower than for F-cMD. Following this trend, the N-terminus in the F-cMD is more rigid than in the F-aMD run. Nevertheless, the N-H vectors of the range 4-8 are quite rigid in both profiles due to the metal-binding Cys5 and Cys8. The order parameter values of F-cMD in the second half of the protein displays more rigidity than in the first half, especially in the metal binding region. Instead, for the F-aMD profile the second half of the protein appears as flexible as the first half, except in the metal binding region that has values similar to the binding region in the first half of the protein. In summary, while the overall dynamics sampled by the F-aMD and F-cMD simulations have some broad global similarities (Figure 6), we could pinpoint differences in the internal motion variations within specific protein regions.
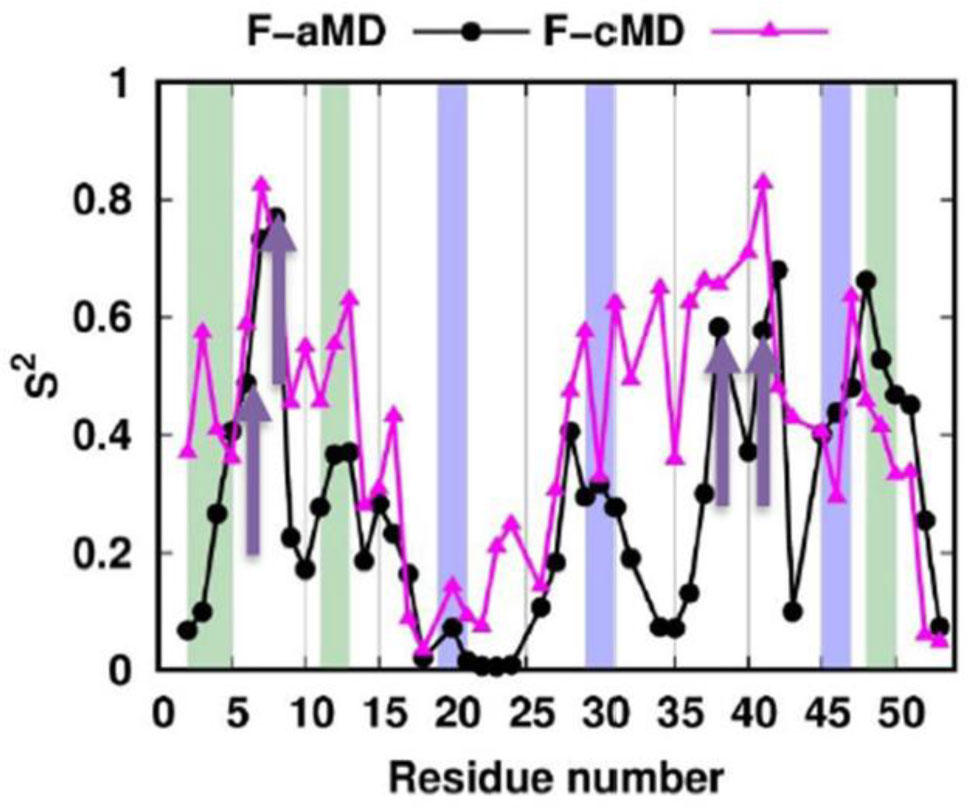 Figure 6. IRED order parameter profiles of the folding simulations. The residues corresponding to β-sheets and 310-helices in the crystal structure are colored with a green and blue background, respectively. The metal binding cysteines are indicated with yellow arrows. The NH vector S2 profiles of the F-aMD and F-cMD trajectories are shown as black and magenta linepoints, respectively.
Figure 6. IRED order parameter profiles of the folding simulations. The residues corresponding to β-sheets and 310-helices in the crystal structure are colored with a green and blue background, respectively. The metal binding cysteines are indicated with yellow arrows. The NH vector S2 profiles of the F-aMD and F-cMD trajectories are shown as black and magenta linepoints, respectively.We analyzed the folding simulations in terms of secondary structures sampled (Figure 7). In the F-cMD conformational ensemble, the second and third strands of the β-sheet are consistently missing, whereas the first strand involves only residues 2 and 3 for the 40% and 1% of the frames, respectively (Figure 7A). The F-aMD trajectory has a higher presence of native β-strands except for residue 2. Regarding helical structures, the F-cMD conformational ensemble typically maintains only two residues of the native first 310-helix and the last residue of the native third helix (Figure 7B). More in detail, residues 20-21 and 47 are in helical conformation in 7% and 37% of the frames, respectively. Also for the helical structures, the F-aMD run shows a better agreement with the crystal. Indeed, all the three helices are sampled at least in 10% of the frames; the third helix in particular is present in more than 60% of the frames. We then conducted a more detailed per-residue analysis of the non-native (i.e. not present in the crystal structure) secondary structures sampled (
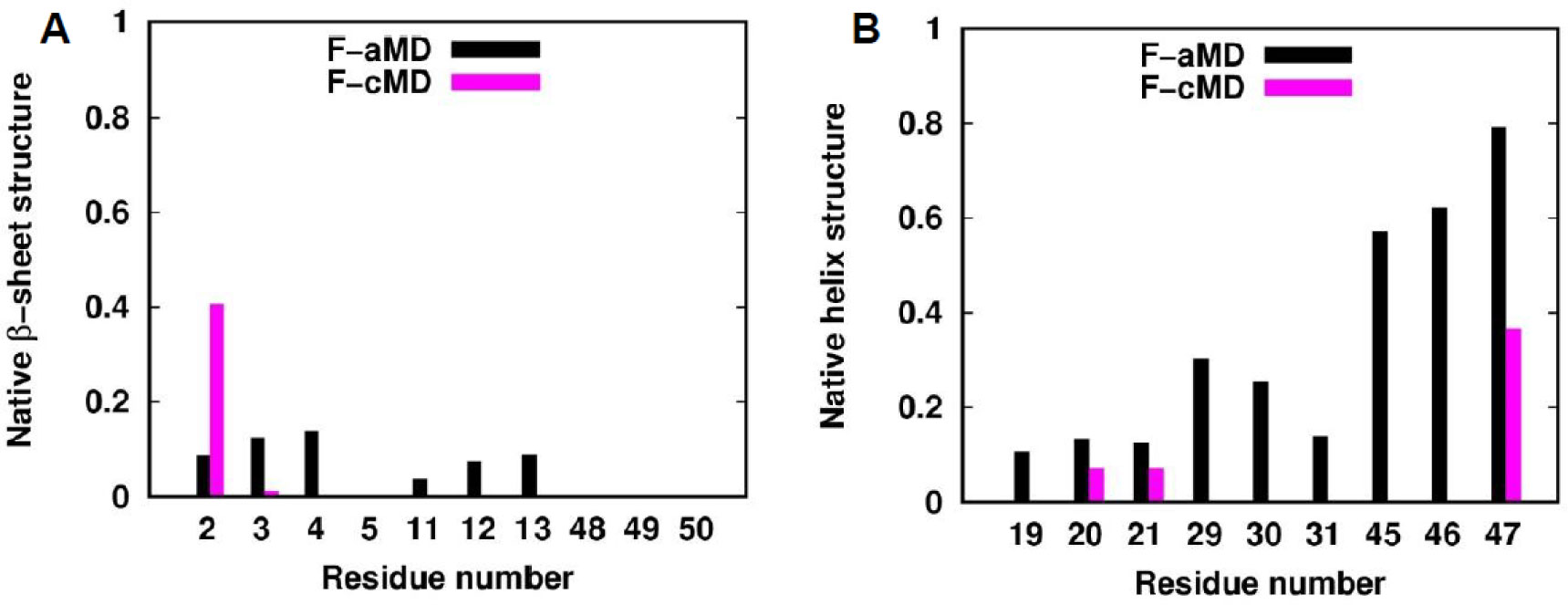 Figure 7. Fraction of native secondary structures sampled in the folding simulations. For each residue being in a secondary structure element in the crystal structure, we report the fraction of conformations that have the same secondary structure in the F-aMD and F-cMD simulations (black and magenta histograms, respectively). (A) Native β-sheets. (B) Native helices.
Figure 7. Fraction of native secondary structures sampled in the folding simulations. For each residue being in a secondary structure element in the crystal structure, we report the fraction of conformations that have the same secondary structure in the F-aMD and F-cMD simulations (black and magenta histograms, respectively). (A) Native β-sheets. (B) Native helices.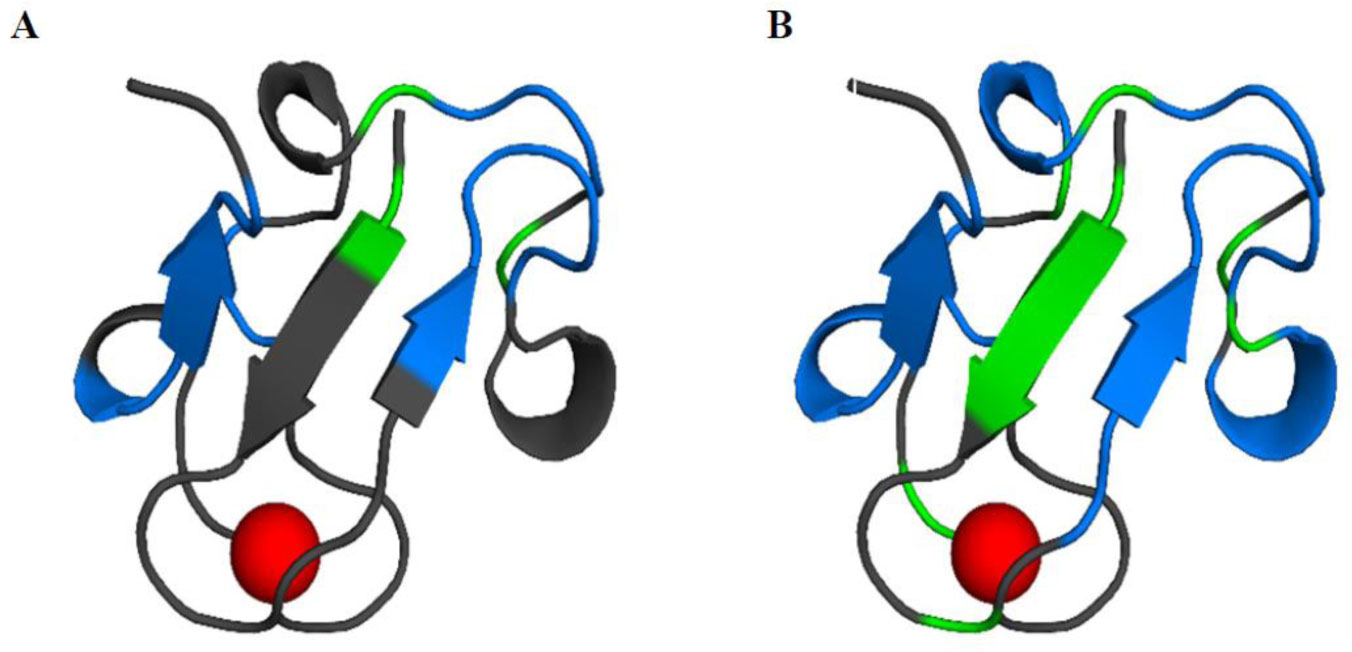 Figure 8. Secondary structures sampled by the F-cMD (A) and F-aMD (B) simulations mapped on the crystal structure of holo-PfRd. The residues where helical or strand structures are sampled in most of the conformations are shown in blue and green, respectively. Unstructured residues are shown in dark gray. The iron ion is shown as a red sphere.
Figure 8. Secondary structures sampled by the F-cMD (A) and F-aMD (B) simulations mapped on the crystal structure of holo-PfRd. The residues where helical or strand structures are sampled in most of the conformations are shown in blue and green, respectively. Unstructured residues are shown in dark gray. The iron ion is shown as a red sphere.Figure 9 displays the correlation of the chemical shifts predicted from the MD trajectories with experimental NMR data for the F-aMD and F-cMD trajectories. As a reference, a completely linearized protein was built in silico and used to compute the same correlation, to be used as a threshold. For the latter system, we observed a high correlation for the Cα atoms, whereas the correlation is poor for both the C and the N atoms. This is not unexpected given the strong dependence of the chemical shifts of the Cα atoms on the aminoacid identity. Both the folding trajectories display a weal correlation for the chemical shifts of the C atoms, with coefficients around 0.41-0.44. The larger variation was observed for the N atoms, with correlation coefficients for the F-aMD, F-cMD and linearized protein of 0.73, 0.70 and 0.68, respectively. Overall, the above data indicate that the F-aMD trajectory has marginally higher correlations with the experimental chemical shifts than the F-cMD trajectory.
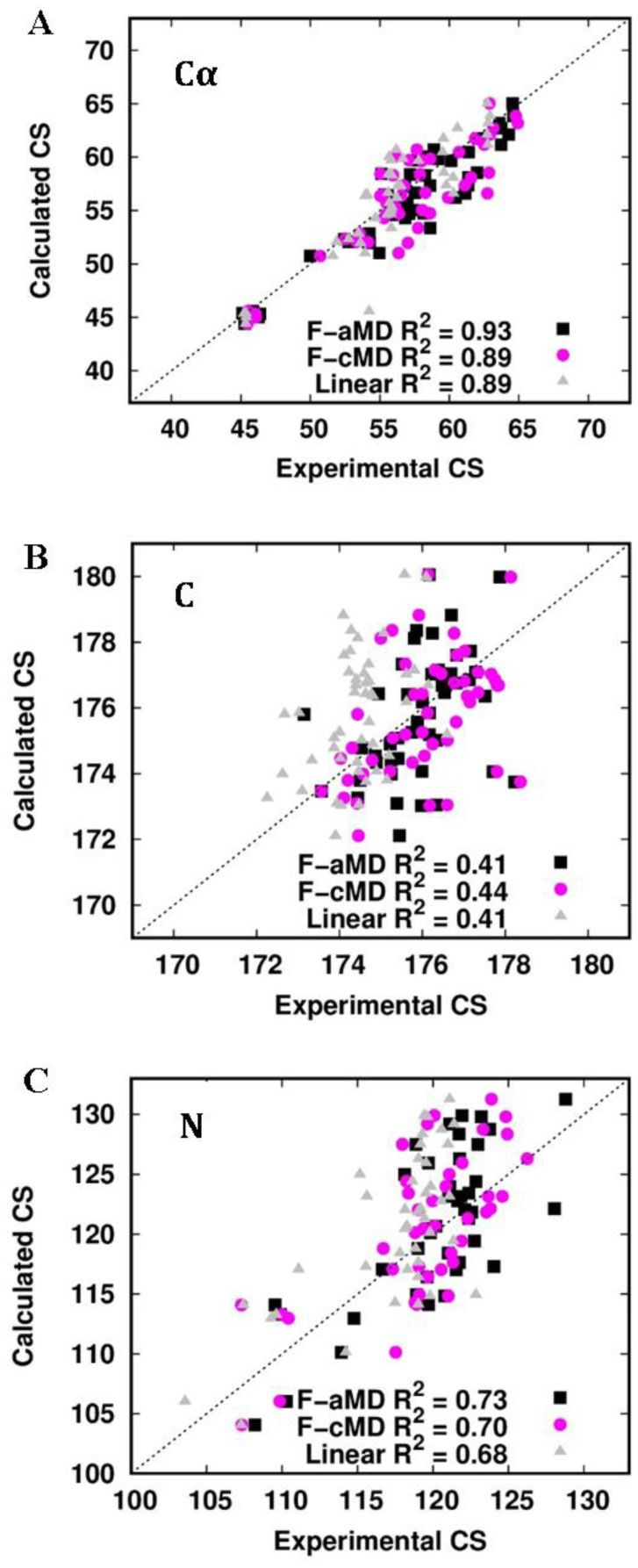 Figure 9. Correlation between experimental and calculated chemical shifts (in ppm) of the F-aMD trajectory, F-cMD trajectory and the linear protein (i.e. a model of the completely linear PfRd). The F-aMD values are shown as black squares. The F-cMD values are shown as magenta circles. The linear protein values are shown as grey triangles. (A) Plot of the Cα atoms chemical shifts. (B) Plot of the C atoms chemical shifts. (C) Plot of the N atoms chemical shifts.
Figure 9. Correlation between experimental and calculated chemical shifts (in ppm) of the F-aMD trajectory, F-cMD trajectory and the linear protein (i.e. a model of the completely linear PfRd). The F-aMD values are shown as black squares. The F-cMD values are shown as magenta circles. The linear protein values are shown as grey triangles. (A) Plot of the Cα atoms chemical shifts. (B) Plot of the C atoms chemical shifts. (C) Plot of the N atoms chemical shifts.The F-aMD simulation has greater flexibility and a better propensity to make native or native-like secondary structures than F-cMD (Figures 7 and 8). In addition, its predicted chemical shifts are slightly closer to the experimental data, although all the folding simulations failed to reproduce the chemical shifts of the C atoms (Figure 9). On the other hand, the APO simulation (starting from the folded structure) features a significant stability of the tertiary structure of the apo-form of PfRd and a strong correlation with the experimental NMR data.
We analyzed the F-aMD trajectory along the simulation time to identify the structural features of possible folding events (Figure 10). The RMSD from the crystal structure of the backbone atoms spans a range from 6.5 to 12.5 Å (Figure 10A). The closest conformation to the crystal structure is reached around 2.2 µs; additional RMSD minima are observed after further 1-1.5 µs. Subsequently, the protein remains almost stable in a plateau at 11 Å for 5 µs. In the second half of the simulation a local minimum at about 7 Å from the crystal is reached three times: around 8, 10.1 and 10.7 µs. The relative partially folded conformations are shown.
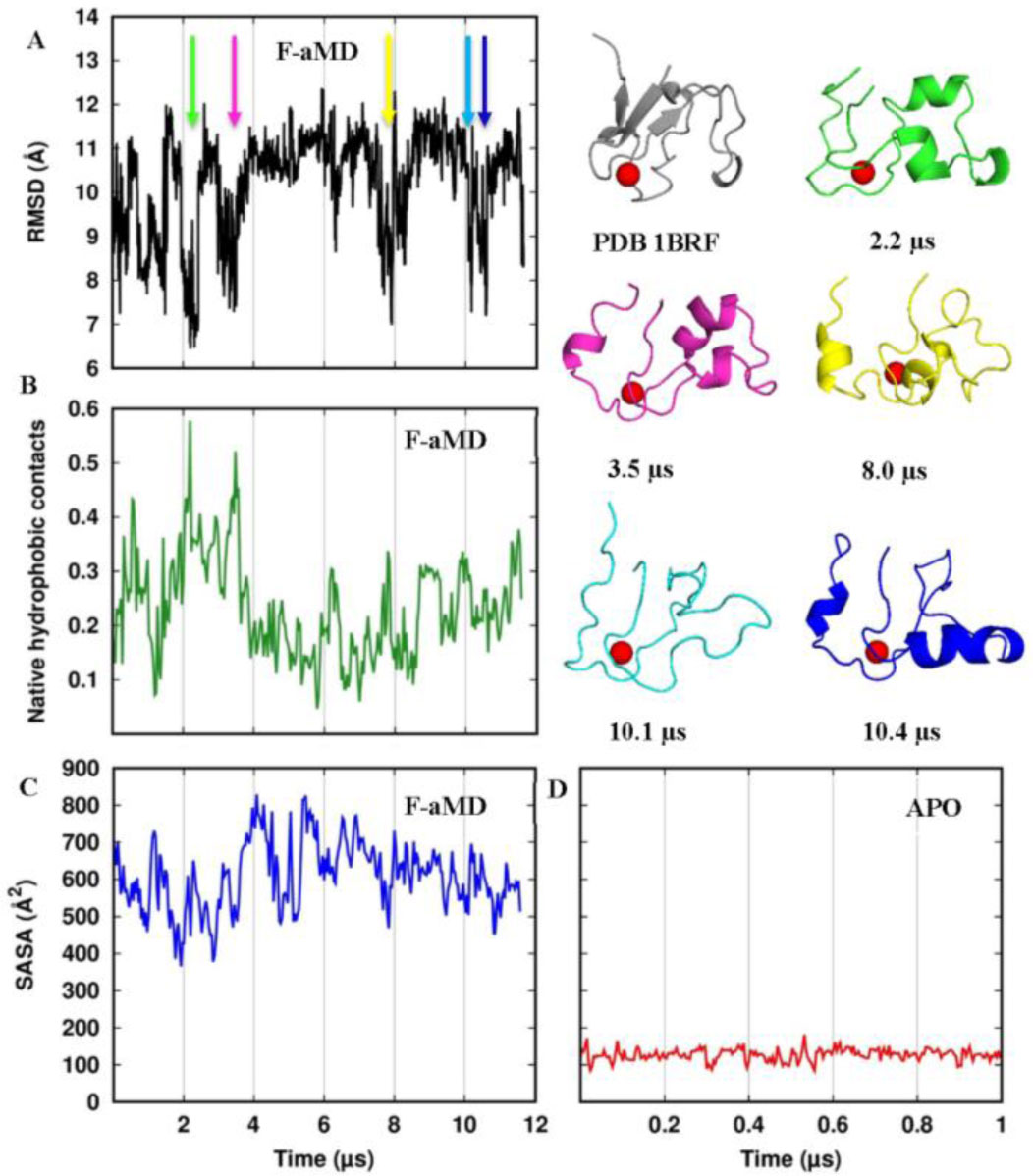 Figure 10. Structural properties along the F-aMD trajectory. (A) backbone RMSD from the crystal structure. The colored arrows indicated minima; the corresponding conformations are shown using the same color. (B) Fraction of native contacts occurring among the side chain residues in the hydrophobic core (residues Trp3, Tyr10, Tyr12, Phe29, Trp36, Phe48, Ile23 and Leu32). (C) SASA of the hydrophobic core. (D) SASA of the hydrophobic core along the APO simulation.
Figure 10. Structural properties along the F-aMD trajectory. (A) backbone RMSD from the crystal structure. The colored arrows indicated minima; the corresponding conformations are shown using the same color. (B) Fraction of native contacts occurring among the side chain residues in the hydrophobic core (residues Trp3, Tyr10, Tyr12, Phe29, Trp36, Phe48, Ile23 and Leu32). (C) SASA of the hydrophobic core. (D) SASA of the hydrophobic core along the APO simulation.On average, hydrophobic residues tend to be in the core of a protein, where solvent accessibility is low, whereas polar residues tend to reside on the surface, where solvent accessibility is high. Thus, to clarify how the packing of the hydrophobic core affects the folding process, we measured the fraction of native hydrophobic contacts among the core residues (i.e. any contact between two hydrophobic residues of the protein core closer than 7 Å and present in the crystal structure). In the F-aMD simulation, the highest fraction of native contacts is reached at 2.2 µs and 3.5 µs, i.e. concurrently with two RMSD minima (Figure 10B). However, even the highest picks on the plot do not exceed the 60% of all the native contacts in the crystal structure. We additionally monitored the Solvent Accessibility Surface Area (SASA) of the PfRd hydrophobic residues as a function of simulation time (Figure 11C). The profile minimum around 400 Å2 is achieved a few ns before 2 µs of simulation. Around 3 µs there is a second minimum. Both these minima occur just before the RMSD minima and native contacts maxima at 2.2 µs and 3.5 µs. These correlations suggest that shielding the hydrophobic core from the solvent is crucial to achieve the folding of the protein. After the first 3 µs, the protein was not able to lower the SASA around 400 Å2 anymore, even though at 8 µs there is another local minimum below 500 Å2, again just some ns before a RMSD minimum. In all the profiles of Figure 10, the best conformation appears at around 2 µs, when the protein first removes the water molecules from the hydrophobic core, and then achieves the highest number of hydrophobic contacts and the most native-like structure (Figure 10, structure in green). This correlation seems to be related to a unique folding event followed by a second similar attempt occurring about 1 µs later.
In general, the correlations among the parameters assessed confirm the strong relationship between the SASA and the number of native hydrophobic contacts profiles (Table 2). In fact, a negative value of −0.51 means that the protein has the necessity of isolating the protein core from the solvent to connect the hydrophobic residues correctly. The correlation of −0.27 between the RMSD from the crystal structure and the fraction of hydrophobic contacts, albeit weak, could indicate the crucial role of the hydrophobic core in pushing the simulation toward correctly folded conformations. The APO simulation constitutes a useful reference to understand the extent to which the folding events in the simulation produced conformations close to the native structure. Thus, considering that the APO trajectory has an average fraction of native hydrophobic contacts in the protein core of 0.74 (data not shown), the F-aMD top value of 0.58 suggests a good attempt but not a perfect packing. In addition, the SASA values sampled in the F-aMD simulation remain significantly larger than the values of the APO simulation, even in correspondence of the two minima (Figure 10C, D). Thus, even if the folding simulation generates a reasonable number of native contacts in the protein core, the hydrophobic residues remain accessible to the water in the conformations explored.
The projection of the F-aMD and APO conformational ensembles on the first two eigenvectors in the Cα space shows the extent of sampling reached by applying the boost potential in contrast to the stability of the APO simulation (
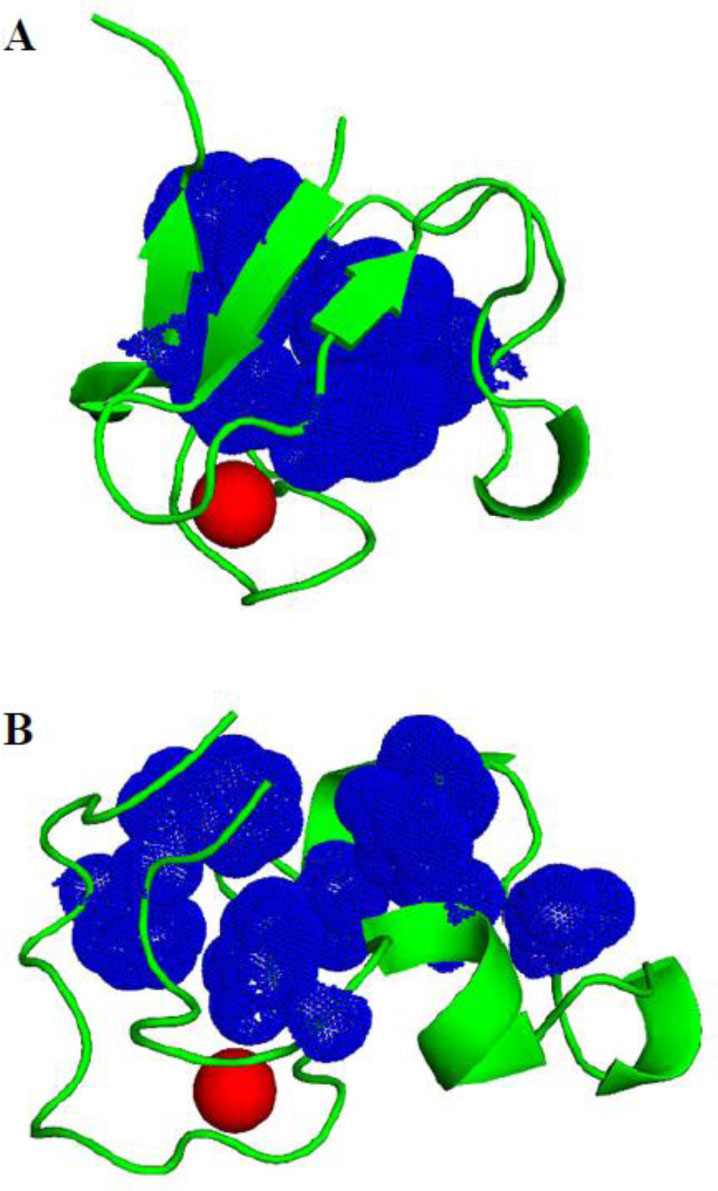 Figure 11. Hydrophobic cluster compactness. The residue side chains of the hydrophobic core are shown as blue dots. The iron ion is shown as red sphere. (A) The highly compact hydrophobic core in the crystal structure PDB 1BRF. (B) The less compact hydrophobic core in the closest to crystal conformation sampled in the F-aMD run.
Figure 11. Hydrophobic cluster compactness. The residue side chains of the hydrophobic core are shown as blue dots. The iron ion is shown as red sphere. (A) The highly compact hydrophobic core in the crystal structure PDB 1BRF. (B) The less compact hydrophobic core in the closest to crystal conformation sampled in the F-aMD run.| Structural parameters compared | R | |
| RMSD | SASA | 0.24 |
| RMSD | Native hydrophobic contacts | −0.27 |
| SASA | Native hydrophobic contacts | −0.51 |
| The strongest correlation is highlighted in bold. R is the Pearson's coefficient. See Figure 10 to observe how each parameter evolves during the simulation. | ||
DownLoad: CSVOur analyses indicate that the APO simulation has sampled conformations close to the folded state, as expected. Nevertheless, there are some visible effects due to the absence of the iron ion on the structural properties of apo-PfRd. In particular, up to three non-native β-sheets between residues in spatial proximity, which partly compensates the degrees of freedom gained with the removal of the iron ion. In addition, we observed that the 310 helical regions tend to assume an H-bonded turn configuration; this is more prominent for the first helix. Similar trends are observed also in the F-aMD simulation, where some non-native β-structures also occur. The occurrence of secondary structures in the F-aMD simulation generally maps with good accuracy to the same residues that are in helical or β-structures also in the crystal structure. This simulation overestimates the occurrence of the elements of helical structure. A significant example is that of the last 310-helix, which extends to involve also residues of the last β-strand of the native sheet (Figure 7). In fact, the full native triple-stranded β-sheet is never sampled, because the simulation is not able to recover all the long-range contacts needed to make the correct β-structures. The NMR chemical shifts of the backbone nuclei of holo-PfRd provide an experimental reference for the understanding of the average dynamic and structural features of the various simulations. For the APO simulation, the agreement of the back-calculated shifts with the experimental data is very good, confirming the absence of significant rearrangements. The situation is somewhat different for the folding simulations, which feature modest correlations for the C and N atoms.
The extensive sampling of the F-aMD trajectory has produced conformations with a compact shape relatively similar to the native structure. This is supported by the analysis of the RMSD from the crystal, and its correlations with the SASA profile and the number of hydrophobic contacts among the core residues (Figure 10 and Table 2). These data suggest that the F-aMD simulation indeed sampled putative folding pathways. In these events, the SASA reduced to 400-500 Å2, and shortly after the RMSD values dropped to about 6-7 Å from the crystal structure. At the same time, the number of native contacts in the hydrophobic core increased to the values observed also in the APO simulation. These results indicate that the major obstacle for the complete folding of holo-PfRd is the residual presence of the solvent molecules in the core residues. As a result, one of the potential driving force in the folding process is weakened. This prevents the compaction of N-terminal part of the structure with the rest of the core (Figure 11). Interestingly, the interaction of the first 15 residues of PfRd with the other parts of the protein has been shown to be an important contributor to the thermostability of the system [20]. An additional contribution limiting our simulation is likely the oversampling of helical structure especially for residues 48-50.
The extensive sampling of one of the smallest known metalloproteins achieved here by a combination of accelerated dynamics and the use of GPGPUs shows that metal-coupled folding is still a challenging task for unbiased MD methods. The main difficulties are not limited to the accuracy of the force field describing the metal binding, such as metal induced protonation/deprotonation [40,41], the polarizable effect [42], the charge transfer [43,44] and multiscale coupling [33,34,35]. Indeed, the standard force field may also induce some bias for the protein part. This is exemplified by the folded apo-form of the protein adopting some non-native secondary structures during the APO simulation. These phenomena affect also the F-aMD and F-cMD simulations. Indeed, there are known limitations in the description of various force fields of interactions that are crucial here, such as those involving phenylalanine side chains [51]. Other authors pointed out that electrostatics and water descriptions could be the weakest force field elements, and proposed that their optimization should consider unfolded proteins [52]. In agreement with this, we suggest that significant methodological work is still needed until unbiased metal-induced folding of metalloproteins can be achieved.
This work was supported by the EUROPEAN COMMISSION (project number 675858, West-Life) and by CIRMMP.
All authors declare no conflict of interest in this paper.
| [1] |
Klepeis JL, Lindorff-Larsen K, Dror RO, et al. (2009) Long-timescale molecular dynamics simulations of protein structure and function. Curr Opin Struct Biol 19: 120–127. doi: 10.1016/j.sbi.2009.03.004

|
| [2] |
Stone JE, Phillips JC, Freddolino PL, et al. (2007) Accelerating molecular modeling applications with graphics processors. J Comput Chem 28: 2618–2640. doi: 10.1002/jcc.20829
|
| [3] |
Perez A, Morrone JA, Simmerling C, et al. (2016) Advances in free-energy-based simulations of protein folding and ligand binding. Curr Opin Struct Biol 36: 25–31. doi: 10.1016/j.sbi.2015.12.002
|
| [4] |
Lane TJ, Shukla D, Beauchamp KA, et al. (2013) To milliseconds and beyond: Challenges in the simulation of protein folding. Curr Opin Struct Biol 23: 58–65. doi: 10.1016/j.sbi.2012.11.002
|
| [5] |
Freddolino PL, Harrison CB, Liu Y, et al. (2010) Challenges in protein-folding simulations. Nat Phys 6: 751–758. doi: 10.1038/nphys1713
|
| [6] |
Best RB (2012) Atomistic molecular simulations of protein folding. Curr Opin Struct Biol 22: 52–61. doi: 10.1016/j.sbi.2011.12.001
|
| [7] | Piana S, Lindorff-Larsen K, Shaw DE (2012) Protein folding kinetics and thermodynamics from atomistic simulation. P Natl Acad Sci USA 109: 17845–17850. |
| [8] |
Suárez E, Lettieri S, Zwier MC, et al. (2014) Simultaneous computation of dynamical and equilibrium information using a weighted ensemble of trajectories. J Chem Theory Comput 10: 2658–2667. doi: 10.1021/ct401065r
|
| [9] |
Pierce LCT, Salomon-Ferrer R, Augusto F. De Oliveira C, et al. (2012) Routine access to millisecond time scale events with accelerated molecular dynamics. J Chem Theory Comput 8: 2997–3002. doi: 10.1021/ct300284c
|
| [10] |
Kubelka J, Hofrichter J, Eaton WA (2004) The protein folding "speed limit." Curr Opin Struct Biol 14: 76–88. doi: 10.1016/j.sbi.2004.01.013
|
| [11] |
Lindorff-Larsen K, Piana S, Dror RO, et al. (2011) How fast-folding proteins fold. Science 334: 517–520. doi: 10.1126/science.1208351
|
| [12] | Putignano V, Rosato A, Banci L, et al. (2018) MetalPDB in 2018: a database of metal sites in biological macromolecular structures. Nucleic Acids Res 41: 459–464. |
| [13] |
Li W, Wang J, Zhang J, et al. (2015) Molecular simulations of metal-coupled protein folding. Curr Opin Struct Biol 30: 25–31. doi: 10.1016/j.sbi.2014.11.006
|
| [14] |
Bentrop D, Bertini I, Iacoviello R, et al. (1999) Structural and dynamical properties of a partially unfolded Fe4S4 protein: Role of the cofactor in protein folding. Biochemistry 38: 4669–4680. doi: 10.1021/bi982647q
|
| [15] |
Blake PR, Summers MF, Park JB, et al. (1991) Determinants of protein hyperthermostability: purification and amino acid sequence of rubredoxin from the hyperthermophilic archaebacterium pyrococcus furiosus and secondary structure of the zinc adduct by NMR. Biochemistry 30: 10885–10895. doi: 10.1021/bi00109a012
|
| [16] | Prakash S, Sundd M, Guptasarma P (2014) The key to the extraordinary thermal stability of P. furiosus holo-rubredoxin: Iron binding-guided packing of a core aromatic cluster responsible for high kinetic stability of the native structure. PLoS One 9: e89703. |
| [17] |
Hernandez G, Jenney FE, Adams MW, et al. (2000) Millisecond time scale conformational flexibility in a hyperthermophile protein at ambient temperature. Proc Natl Acad Sci USA 97: 3166–3170. doi: 10.1073/pnas.97.7.3166
|
| [18] | Rader AJ (2010) Thermostability in rubredoxin and its relationship to mechanical rigidity. Phys Biol 7: 016002. |
| [19] |
Bonomi F, Iametti S, Ferranti P, et al. (2008) "Iron priming" guides folding of denatured aporubredoxins. J Biol Inorg Chem 13: 981–991. doi: 10.1007/s00775-008-0385-4
|
| [20] |
Zartler ER, Jenney FE, Terrell M, et al. (2001) Structural basis for thermostability in aporubredoxins from Pyrococcus furiosus and Clostridium pasteurianum. Biochemistry 40: 7279–7290. doi: 10.1021/bi0026831
|
| [21] |
Cavagnero S, Debe DA, Zhou ZH, et al. (1998) Kinetic role of electrostatic interactions in the unfolding of hyperthermophilic and mesophilic rubredoxins. Biochemistry 37: 3369–3376. doi: 10.1021/bi9721795
|
| [22] |
Strop P, Mayo SL (1999) Rubredoxin variant folds without iron. J Am Chem Soc 121: 2341–2345. doi: 10.1021/ja9834780
|
| [23] |
Hamelberg D, Mongan J, McCammon JA (2004) Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J Chem Phys 120: 11919–11929. doi: 10.1063/1.1755656
|
| [24] |
Doshi U, Hamelberg D (2015) Towards fast, rigorous and efficient conformational sampling of biomolecules: Advances in accelerated molecular dynamics. BBA-Gen Subjects 1850: 878–888. doi: 10.1016/j.bbagen.2014.08.003
|
| [25] |
Miao Y, Feixas F, Eun C (2015) Accelerated molecular dynamics simulations of protein folding. J Comput Chem 36: 1536–1549. doi: 10.1002/jcc.23964
|
| [26] | Case DA, Cerutti DS, Cheatham TE, et al. (2017) Amber 2017, University of California, San Francisco. |
| [27] |
Carvalho ATP, Teixeira AFS, Ramos MJ (2013) Parameters for molecular dynamics simulations of iron-sulfur proteins. J Comput Chem 34: 1540–1548. doi: 10.1002/jcc.23287
|
| [28] |
Bertini I, Case DA, Ferella L, et al. (2011) A grid-enabled web portal for NMR structure refinement with AMBER. Bioinformatics 27: 2384–2390. doi: 10.1093/bioinformatics/btr415
|
| [29] |
Wassenaar TA, van Dijk M, Loureiro-Ferreira N, et al. (2012) WeNMR: Structural biology on the grid. J Grid Comput 10: 743–767. doi: 10.1007/s10723-012-9246-z
|
| [30] |
Prompers JJ, Brüschweiler R, Bruschweiler R (2002) General framework for studying the dynamics of folded and nonfolded proteins by NMR relaxation spectroscopy and MD simulation. J Am Chem Soc 124: 4522–4534. doi: 10.1021/ja012750u
|
| [31] |
Korzhnev DM, Billeter M, Arseniev AS, et al. (2001) NMR studies of Brownian tumbling and internal motions in proteins. Prog Nucl Mag Res Sp 38: 197–266. doi: 10.1016/S0079-6565(00)00028-5
|
| [32] |
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22: 2577–2637. doi: 10.1002/bip.360221211
|
| [33] |
Rost B, Sander C (1993) Prediction of protein secondary structure at better than 70% accuracy. J Mol Biol 232: 584–599. doi: 10.1006/jmbi.1993.1413
|
| [34] |
Li DW, Brüschweiler R (2012) PPM: A side-chain and backbone chemical shift predictor for the assessment of protein conformational ensembles. J Biomol NMR 54: 257–265. doi: 10.1007/s10858-012-9668-8
|
| [35] |
Hiller R, Zhou ZH, Adams MW, et al. (1997) Stability and dynamics in a hyperthermophilic protein with melting temperature close to 200 degrees C. Proc Natl Acad Sci USA 94: 11329–11332. doi: 10.1073/pnas.94.21.11329
|
| [36] |
Ishima R, Torchia DA (2000) Protein dynamics from NMR. Nat Struct Biol 7: 740–743. doi: 10.1038/78963
|
| [37] |
Jarymowycz VA, Stone MJ (2006) Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem Rev 106: 1624–1671. doi: 10.1021/cr040421p
|
| [38] |
LeMaster DM (1999) NMR relaxation order parameter analysis of the dynamics of protein side chains. J Am Chem Soc 121: 1726–1742. doi: 10.1021/ja982988r
|
| [39] |
Ruschak AM, Kay LE (2010) Methyl groups as probes of supra-molecular structure, dynamics and function. J Biomol NMR 46: 75–87. doi: 10.1007/s10858-009-9376-1
|
| [40] |
Bougault CM, Eidsness MK, Prestegard JH (2003) Hydrogen bonds in rubredoxins from mesophilic and hyperthermophilic organisms. Biochemistry 42: 4357–4372. doi: 10.1021/bi027264d
|
| [41] |
Prestegard JH, Bougault CM, Kishore AI (2004) Residual dipolar couplings in structure determination of biomolecules. Chem Rev 104: 3519–3540. doi: 10.1021/cr030419i
|
| [42] |
Cho-Chung YS, Pitot HC (1968) Regulatory effects of nicotinamide on tryptophan pyrrolase synthesis in rat liver in vivo. Eur J Biochem 3: 401–406. doi: 10.1111/j.1432-1033.1967.tb19543.x
|
| [43] |
Blasie CA, Berg JM (2002) Structur e-based thermodynamic analysis of a coupled metal binding-protein folding reaction involving a zinc finger peptide. Biochemistry 41: 15068–15073. doi: 10.1021/bi026621h
|
| [44] |
Weinkam P, Romesberg FE, Wolynes PG (2009) Chemical frustration in the protein folding landscape: Grand canonical ensemble simulations of cytochrome c. Biochemistry 48: 2394–2402. doi: 10.1021/bi802293m
|
| [45] |
Devereux M, Gresh N, Piquemal JP, et al. (2014) A supervised fitting approach to force field parametrization with application to the SIBFA polarizable force field. J Comput Chem 35: 1577–1591. doi: 10.1002/jcc.23661
|
| [46] |
Wu R, Lu Z, Cao Z, et al. (2011) A transferable nonbonded pairwise force field to model zinc interactions in metalloproteins. J Chem Theory Comput 7: 433–443. doi: 10.1021/ct100525r
|
| [47] |
Sakharov DV, Lim C (2005) Zn protein simulations including charge transfer and local polarization effects. J Am Chem Soc 127: 4921–4929. doi: 10.1021/ja0429115
|
| [48] |
Chakravorty DK, Wang B, Lee CW, et al. (2012) Simulations of allosteric motions in the zinc sensor CzrA. J Am Chem Soc 134: 3367–3376. doi: 10.1021/ja208047b
|
| [49] |
Chakravorty DK, Parker TM, Guerra AJ, et al. (2013) Energetics of zinc-mediated interactions in the allosteric pathways of metal sensor proteins. J Am Chem Soc 135: 30–33. doi: 10.1021/ja309170g
|
| [50] |
Reyes-Caballero H, Campanello GC, Giedroc DP (2011) Metalloregulatory proteins: Metal selectivity and allosteric switching. Biophys Chem 156: 103–114. doi: 10.1016/j.bpc.2011.03.010
|
| [51] |
Andrews CT, Elcock AH (2013) Molecular dynamics simulations of highly crowded amino acid solutions: comparisons of eight different force field combinations with experiment and with each other. J Chem Theory Comput 9: 4585–4602. doi: 10.1021/ct400371h
|
| [52] |
Abriata LA, Dal Peraro M (2015) Assessing the potential of atomistic molecular dynamics simulations to probe reversible protein-protein recognition and binding. Sci Rep 5: 10549. doi: 10.1038/srep10549
|
 biophy-05-01-077-s1.pdf biophy-05-01-077-s1.pdf |
 |
| 1. | Davide Sala, Ugo Cosentino, Anna Ranaudo, Claudio Greco, Giorgio Moro, Dynamical Behavior and Conformational Selection Mechanism of the Intrinsically Disordered Sic1 Kinase-Inhibitor Domain, 2020, 10, 2075-1729, 110, 10.3390/life10070110 | |
| 2. | Marzieh Gharouni, Hamid Mosaddeghi, Jamshid Mehrzad, Ali Es-haghi, Alireza Motavalizadehkakhky, In silico profiling and structural insights of zinc metal ion on O6-methylguanine methyl transferase and its interactions using molecular dynamics approach, 2021, 27, 1610-2940, 10.1007/s00894-020-04631-x | |
| 3. | Xiaojuan Hu, Maja-Olivia Lenz-Himmer, Carsten Baldauf, Better force fields start with better data: A data set of cation dipeptide interactions, 2022, 9, 2052-4463, 10.1038/s41597-022-01297-3 | |
| 4. | Christina Eleftheria Tzeliou, Markella Aliki Mermigki, Demeter Tzeli, Review on the QM/MM Methodologies and Their Application to Metalloproteins, 2022, 27, 1420-3049, 2660, 10.3390/molecules27092660 | |
| 5. | Francis E. Jenney, Hongxin Wang, Simon J. George, Jin Xiong, Yisong Guo, Leland B. Gee, Juan José Marizcurrena, Susana Castro-Sowinski, Anna Staskiewicz, Yoshitaka Yoda, Michael Y. Hu, Kenji Tamasaku, Nobumoto Nagasawa, Lei Li, Hiroaki Matsuura, Tzanko Doukov, Stephen P. Cramer, Temperature-dependent iron motion in extremophile rubredoxins – no need for ‘corresponding states’, 2024, 14, 2045-2322, 10.1038/s41598-024-62261-2 | |
| 6. | Karuna Anna Sajeevan, Bibek Acharya, Sakib Ferdous, Dan M. Park, Joseph A. Cotruvo, Ratul Chowdhury, Computationally Derived Structural Insights into Rare Earth Selectivity in Lanmodulin and its Variants, 2025, 20010370, 10.1016/j.csbj.2025.02.005 |











Figures(11) / Tables(2)

Davide Sala, Andrea Giachetti, Antonio Rosato. Molecular dynamics simulations of metalloproteins: A folding study of rubredoxin from Pyrococcus furiosus[J]. AIMS Biophysics, 2018, 5(1): 77-96. doi: 10.3934/biophy.2018.1.77

| Type of simulation | Simulation length (µs) | Starting structure |
| Classical-Apo (APO) | 1 | Crystal structure without metal ion |
| Folding Accelerated (F-aMD) | 11.6 | Unfolded state with the metal bound |
| Folding Classical (F-cMD) | 1 | Unfolded state with the metal bound |
| The acronyms used in the text to identify each simulation are given in brackets in the first column. Both folding runs started from the same elongated conformation with the iron ion bound. | ||
DownLoad: CSV| Structural parameters compared | R | |
| RMSD | SASA | 0.24 |
| RMSD | Native hydrophobic contacts | −0.27 |
| SASA | Native hydrophobic contacts | −0.51 |
| The strongest correlation is highlighted in bold. R is the Pearson's coefficient. See Figure 10 to observe how each parameter evolves during the simulation. | ||
DownLoad: CSV| Type of simulation | Simulation length (µs) | Starting structure |
| Classical-Apo (APO) | 1 | Crystal structure without metal ion |
| Folding Accelerated (F-aMD) | 11.6 | Unfolded state with the metal bound |
| Folding Classical (F-cMD) | 1 | Unfolded state with the metal bound |
| The acronyms used in the text to identify each simulation are given in brackets in the first column. Both folding runs started from the same elongated conformation with the iron ion bound. | ||
| Structural parameters compared | R | |
| RMSD | SASA | 0.24 |
| RMSD | Native hydrophobic contacts | −0.27 |
| SASA | Native hydrophobic contacts | −0.51 |
| The strongest correlation is highlighted in bold. R is the Pearson's coefficient. See Figure 10 to observe how each parameter evolves during the simulation. | ||





