Citation: Oleg A. Karpov, Gareth W. Fearnley, Gina A. Smith, Jayakanth Kankanala, Michael J. McPherson, Darren C. Tomlinson, Michael A. Harrison, Sreenivasan Ponnambalam. Receptor tyrosine kinase structure and function in health and disease[J]. AIMS Biophysics, 2015, 2(4): 476-502. doi: 10.3934/biophy.2015.4.476

| [1] |
Cohen S (1965) The stimulation of epidermal proliferation by a specific protein (EGF). Dev Biol 12: 394–407. doi: 10.1016/0012-1606(65)90005-9

|
| [2] |
Cohen S, Carpenter G, Lembach KJ (1975) Interaction of epidermal growth factor (EGF) with cultured fibroblasts. Adv Metab Disord 8: 265–284. doi: 10.1016/B978-0-12-027308-9.50024-X
|
| [3] |
Carpenter G, King L Jr., Cohen S (1978) Epidermal growth factor stimulates phosphorylation in membrane preparations in vitro. Nature 276: 409–410. doi: 10.1038/276409a0
|
| [4] |
Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141: 1117–1134. doi: 10.1016/j.cell.2010.06.011
|
| [5] |
Lemmon MA, Schlessinger J, Ferguson KM (2014) The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb Perspect Biol 6: a020768. doi: 10.1101/cshperspect.a020768
|
| [6] | Kovacs E, Zorn JA, Huang Y, et al. (2015) A Structural Perspective on the Regulation of the Epidermal Growth Factor Receptor. Annu Rev Biochem. E-pub ahead of print. |
| [7] |
Ullrich A, Schlessinger J (1990) Signal transduction by receptors with tyrosine kinase activity. Cell 61: 203–212. doi: 10.1016/0092-8674(90)90801-K
|
| [8] |
Moriki T, Maruyama H, Maruyama IN (2001) Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J Mol Biol 311: 1011–1026. doi: 10.1006/jmbi.2001.4923
|
| [9] | Burgess AW, Cho HS, Eigenbrot C, et al. (2003) An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell 12: 541–552. |
| [10] | Yokoyama N, Miller WT (2003) Biochemical properties of the Cdc42-associated tyrosine kinase ACK1. Substrate specificity, authphosphorylation, and interaction with Hck. J Biol Chem 278: 47713–47723. |
| [11] |
Burgess AW (2008) EGFR family: structure physiology signalling and therapeutic targets. Growth Factors 26: 263–274. doi: 10.1080/08977190802312844
|
| [12] |
Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2: 127–137. doi: 10.1038/35052073
|
| [13] |
Jura N, Shan Y, Cao X, et al. (2009) Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc Natl Acad Sci USA 106: 21608–21613. doi: 10.1073/pnas.0912101106
|
| [14] |
Endres NF, Engel K, Das R, et al. (2011) Regulation of the catalytic activity of the EGF receptor. Curr Opin Struct Biol 21: 777–784. doi: 10.1016/j.sbi.2011.07.007
|
| [15] |
Li S, Schmitz KR, Jeffrey PD, et al. (2005) Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 7: 301–311. doi: 10.1016/j.ccr.2005.03.003
|
| [16] |
Vecchi M, Carpenter G (1997) Constitutive proteolysis of the ErbB-4 receptor tyrosine kinase by a unique, sequential mechanism. J Cell Biol 139: 995–1003. doi: 10.1083/jcb.139.4.995
|
| [17] | Codony-Servat J, Albanell J, Lopez-Talavera JC, et al. (1999) Cleavage of the HER2 ectodomain is a pervanadate-activable process that is inhibited by the tissue inhibitor of metalloproteases-1 in breast cancer cells. Cancer Res 59: 1196–1201. |
| [18] |
Ullrich A, Coussens L, Hayflick JS, et al. (1984) Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 309: 418–425. doi: 10.1038/309418a0
|
| [19] | Gamou S, Shimizu N (1988) Glycosylation of the epidermal growth factor receptor and its relationship to membrane transport and ligand binding. J Biochem 104: 388–396. |
| [20] | Terman BI, Carrion ME, Kovacs E, et al. (1991) Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene 6: 1677–1683. |
| [21] |
Holmes K, Roberts OL, Thomas AM, et al. (2007) Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cellular Signalling 19: 2003–2012. doi: 10.1016/j.cellsig.2007.05.013
|
| [22] | Waltenberger J, Claesson-Welsh L, Siegbahn A, et al. (1994) Different signal transduction properties of KDR and Flt1, 2 receptors for vascular endothelial growth factor. J Biol Chem 269: 26988–26995. |
| [23] | Koch S, Claesson-Welsh L (2012) Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med 2: a006502. |
| [24] |
Takahashi T, Shibuya M (1997) The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-gamma pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene 14: 2079–2089. doi: 10.1038/sj.onc.1201047
|
| [25] |
Shinkai A, Ito M, Anazawa H, et al. (1998) Mapping of the sites involved in ligand association and dissociation at the extracellular domain of the kinase insert domain-containing receptor for vascular endothelial growth factor. J Biol Chem 273: 31283–31288. doi: 10.1074/jbc.273.47.31283
|
| [26] |
Ruch C, Skiniotis G, Steinmetz MO, et al. (2007) Structure of a VEGF-VEGF receptor complex determined by electron microscopy. Nat Struct Mol Biol 14: 249–250. doi: 10.1038/nsmb1202
|
| [27] |
Toffalini F, Demoulin JB (2010) New insights into the mechanisms of hematopoietic cell transformation by activated receptor tyrosine kinases. Blood 116: 2429–2437. doi: 10.1182/blood-2010-04-279752
|
| [28] |
Yang Y, Yuzawa S, Schlessinger J (2008) Contacts between membrane proximal regions of the PDGF receptor ectodomain are required for receptor activation but not for receptor dimerization. Proc Natl Acad Sci U S A 105: 7681–7686. doi: 10.1073/pnas.0802896105
|
| [29] |
De Meyts P, Whittaker J (2002) Structural biology of insulin and IGF1 receptors: implications for drug design. Nat Rev Drug Discov 1: 769–783. doi: 10.1038/nrd917
|
| [30] |
Heldin CH, Ostman A (1996) Ligand-induced dimerization of growth factor receptors: variations on the theme. Cytokine Growth Factor Rev 7: 3–10. doi: 10.1016/1359-6101(96)00002-0
|
| [31] |
Ward CW, Lawrence MC (2009) Ligand-induced activation of the insulin receptor: a multi-step process involving structural changes in both the ligand and the receptor. Bioessays 31: 422–434. doi: 10.1002/bies.200800210
|
| [32] | Soos MA, Field CE, Siddle K (1993) Purified hybrid insulin/insulin-like growth factor-I receptors bind insulin-like growth factor-I, but not insulin, with high affinity. Biochem J 290 ( Pt 2): 419–426. |
| [33] |
Xu H, Abe T, Liu JK, et al. (2014) Normal activation of discoidin domain receptor 1 mutants with disulfide cross-links, insertions, or deletions in the extracellular juxtamembrane region: mechanistic implications. J Biol Chem 289: 13565–13574. doi: 10.1074/jbc.M113.536144
|
| [34] |
Mason I (2007) Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nat Rev Neurosci 8: 583–596. doi: 10.1038/nrn2189
|
| [35] |
Mohammadi M, Olsen SK, Ibrahimi OA (2005) Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev 16: 107–137. doi: 10.1016/j.cytogfr.2005.01.008
|
| [36] |
Grunewald FS, Prota AE, Giese A, et al. (2010) Structure-function analysis of VEGF receptor activation and the role of coreceptors in angiogenic signaling. Biochim Biophys Acta 1804: 567–580. doi: 10.1016/j.bbapap.2009.09.002
|
| [37] |
Nolen B, Taylor S, Ghosh G (2004) Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell 15: 661–675. doi: 10.1016/j.molcel.2004.08.024
|
| [38] |
Hubbard SR (2004) Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol 5: 464–471. doi: 10.1038/nrm1399
|
| [39] |
Huse M, Kuriyan J (2002) The conformational plasticity of protein kinases. Cell 109: 275–282. doi: 10.1016/S0092-8674(02)00741-9
|
| [40] |
Wybenga-Groot LE, Baskin B, Ong SH, et al. (2001) Structural basis for autoinhibition of the Ephb2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell 106: 745–757. doi: 10.1016/S0092-8674(01)00496-2
|
| [41] |
Mol CD, Dougan DR, Schneider TR, et al. (2004) Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem 279: 31655–31663. doi: 10.1074/jbc.M403319200
|
| [42] |
Till JH, Becerra M, Watty A, et al. (2002) Crystal structure of the MuSK tyrosine kinase: insights into receptor autoregulation. Structure 10: 1187–1196. doi: 10.1016/S0969-2126(02)00814-6
|
| [43] |
Griffith J, Black J, Faerman C, et al. (2004) The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell 13: 169–178. doi: 10.1016/S1097-2765(03)00505-7
|
| [44] |
Dibb NJ, Dilworth SM, Mol CD (2004) Switching on kinases: oncogenic activation of BRAF and the PDGFR family. Nat Rev Cancer 4: 718–727. doi: 10.1038/nrc1434
|
| [45] | Heisermann GJ, Wiley HS, Walsh BJ, et al. (1990) Mutational removal of the Thr669 and Ser671 phosphorylation sites alters substrate specificity and ligand-induced internalization of the epidermal growth factor receptor. J Biol Chem 265: 12820–12827. |
| [46] |
Welsh JB, Gill GN, Rosenfeld MG, et al. (1991) A negative feedback loop attenuates EGF-induced morphological changes. J Cell Biol 114: 533–543. doi: 10.1083/jcb.114.3.533
|
| [47] |
Hsu SC, Hung MC (2007) Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J Biol Chem 282: 10432–10440. doi: 10.1074/jbc.M610014200
|
| [48] |
Red Brewer M, Choi SH, Alvarado D, et al. (2009) The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell 34: 641–651. doi: 10.1016/j.molcel.2009.04.034
|
| [49] |
Jura N, Endres NF, Engel K, et al. (2009) Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 137: 1293–1307. doi: 10.1016/j.cell.2009.04.025
|
| [50] |
Favelyukis S, Till JH, Hubbard SR, et al. (2001) Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol 8: 1058–1063. doi: 10.1038/nsb721
|
| [51] |
Koch S, Tugues S, Li X, et al. (2011) Signal transduction by vascular endothelial growth factor receptors. Biochem J 437: 169–183. doi: 10.1042/BJ20110301
|
| [52] |
Roskoski R Jr (2007) Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol Hematol 62: 179–213. doi: 10.1016/j.critrevonc.2007.01.006
|
| [53] |
Kawamura H, Li X, Harper SJ, et al. (2008) Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res 68: 4683–4692. doi: 10.1158/0008-5472.CAN-07-6577
|
| [54] |
Harper SJ, Bates DO (2008) VEGF-A splicing: the key to anti-angiogenic therapeutics? Nature Rev Cancer 8: 880–887. doi: 10.1038/nrc2505
|
| [55] |
Zhang X, Gureasko J, Shen K, et al. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125: 1137–1149. doi: 10.1016/j.cell.2006.05.013
|
| [56] |
Knowles PP, Murray-Rust J, Kjaer S, et al. (2006) Structure and chemical inhibition of the RET tyrosine kinase domain. J Biol Chem 281: 33577–33587. doi: 10.1074/jbc.M605604200
|
| [57] |
Pawson T (2004) Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116: 191–203. doi: 10.1016/S0092-8674(03)01077-8
|
| [58] | Schlessinger J, Lemmon MA (2003) SH2 and PTB domains in tyrosine kinase signaling. Sci STKE 2003: RE12. |
| [59] |
Eswarakumar VP, Lax I, Schlessinger J (2005) Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 16: 139–149. doi: 10.1016/j.cytogfr.2005.01.001
|
| [60] |
Seet BT, Dikic I, Zhou MM, et al. (2006) Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol 7: 473–483. doi: 10.1038/nrm1960
|
| [61] |
Ladbury JE, Arold S (2000) Searching for specificity in SH domains. Chem Biol 7: R3–8. doi: 10.1016/S1074-5521(00)00067-3
|
| [62] |
Eck MJ, Pluskey S, Trub T, et al. (1996) Spatial constraints on the recognition of phosphoproteins by the tandem SH2 domains of the phosphatase SH-PTP2. Nature 379: 277–280. doi: 10.1038/379277a0
|
| [63] |
Pawson T (2007) Dynamic control of signaling by modular adaptor proteins. Curr Opin Cell Biol 19: 112–116. doi: 10.1016/j.ceb.2007.02.013
|
| [64] |
Songyang Z, Shoelson SE, Chaudhuri M, et al. (1993) SH2 domains recognize specific phosphopeptide sequences. Cell 72: 767–778. doi: 10.1016/0092-8674(93)90404-E
|
| [65] |
Waksman G, Shoelson SE, Pant N, et al. (1993) Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell 72: 779–790. doi: 10.1016/0092-8674(93)90405-F
|
| [66] |
Rhee SG (2001) Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem 70: 281–312. doi: 10.1146/annurev.biochem.70.1.281
|
| [67] |
Chattopadhyay A, Vecchi M, Ji Q, et al. (1999) The role of individual SH2 domains in mediating association of phospholipase C-gamma1 with the activated EGF receptor. J Biol Chem 274: 26091–26097. doi: 10.1074/jbc.274.37.26091
|
| [68] |
Bae JH, Lew ED, Yuzawa S, et al. (2009) The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell 138: 514–524. doi: 10.1016/j.cell.2009.05.028
|
| [69] |
Marshall CJ (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80: 179–185. doi: 10.1016/0092-8674(95)90401-8
|
| [70] |
Kitano H (2004) Biological robustness. Nat Rev Genet 5: 826–837. doi: 10.1038/nrg1471
|
| [71] |
Kholodenko BN (2006) Cell-signalling dynamics in time and space. Nat Rev Mol Cell Biol 7: 165–176. doi: 10.1038/nrm1838
|
| [72] |
Ostman A, Bohmer FD (2001) Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol 11: 258–266. doi: 10.1016/S0962-8924(01)01990-0
|
| [73] | Bae YS, Kang SW, Seo MS, et al. (1997) Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem 272: 217–221. |
| [74] | Buday L, Warne PH, Downward J (1995) Downregulation of the Ras activation pathway by MAP kinase phosphorylation of Sos. Oncogene 11: 1327–1331. |
| [75] |
Mittar S, Ulyatt C, Howell GJ, et al. (2009) VEGFR1 receptor tyrosine kinase localization to the Golgi apparatus is calcium-dependent. Exp Cell Res 315: 877–889. doi: 10.1016/j.yexcr.2008.12.020
|
| [76] | Goh LK, Sorkin A (2013) Endocytosis of receptor tyrosine kinases. Cold Spring Harb Perspect Biol 5: a017459. |
| [77] |
Sorkin A, Goh LK (2009) Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res 315: 683–696. doi: 10.1016/j.yexcr.2008.07.029
|
| [78] |
Auciello G, Cunningham DL, Tatar T, et al. (2013) Regulation of fibroblast growth factor receptor signalling and trafficking by Src and Eps8. J Cell Sci 126: 613–624. doi: 10.1242/jcs.116228
|
| [79] |
Ewan LC, Jopling HM, Jia H, et al. (2006) Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic 7: 1270–1282. doi: 10.1111/j.1600-0854.2006.00462.x
|
| [80] |
Sigismund S, Woelk T, Puri C, et al. (2005) Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci USA 102: 2760–2765. doi: 10.1073/pnas.0409817102
|
| [81] |
Bruns AF, Herbert SP, Odell AF, et al. (2010) Ligand-stimulated VEGFR2 signaling is regulated by co-ordinated trafficking and proteolysis. Traffic 11: 161–174. doi: 10.1111/j.1600-0854.2009.01001.x
|
| [82] |
Henriksen L, Grandal MV, Knudsen SL, et al. (2013) Internalization mechanisms of the epidermal growth factor receptor after activation with different ligands. PLoS One 8: e58148. doi: 10.1371/journal.pone.0058148
|
| [83] |
Miaczynska M (2013) Effects of membrane trafficking on signaling by receptor tyrosine kinases. Cold Spring Harb Perspect Biol 5: a009035. doi: 10.1101/cshperspect.a009035
|
| [84] | de Melker AA, van der Horst G, Calafat J, et al. (2001) c-Cbl ubiquitinates the EGF receptor at the plasma membrane and remains receptor associated throughout the endocytic route. J Cell Sci 114: 2167–2178. |
| [85] | Duval M, Bedard-Goulet S, Delisle C, et al. (2003) Vascular endothelial growth factor-dependent down-regulation of Flk-1/KDR involves Cbl-mediated ubiquitination. Consequences on nitric oxide production from endothelial cells. J Biol Chem 278: 20091–20097. |
| [86] |
Citri A, Alroy I, Lavi S, et al. (2002) Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J 21: 2407–2417. doi: 10.1093/emboj/21.10.2407
|
| [87] |
Xu Q (2002) Role of heat shock proteins in atherosclerosis. Arterioscler Thromb Vasc Biol 22: 1547–1559. doi: 10.1161/01.ATV.0000029720.59649.50
|
| [88] |
Ehrlich ES, Wang T, Luo K, et al. (2009) Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc Natl Acad Sci USA 106: 20330–20335. doi: 10.1073/pnas.0810571106
|
| [89] |
Bruns AF, Yuldasheva N, Latham AM, et al. (2012) A heat-shock protein axis regulates VEGFR2 proteolysis, blood vessel development and repair. PLoS One 7: e48539. doi: 10.1371/journal.pone.0048539
|
| [90] |
Samant RS, Clarke PA, Workman P (2014) E3 ubiquitin ligase Cullin-5 modulates multiple molecular and cellular responses to heat shock protein 90 inhibition in human cancer cells. Proc Natl Acad Sci U S A 111: 6834–6839. doi: 10.1073/pnas.1322412111
|
| [91] |
Ascano M, Richmond A, Borden P, et al. (2009) Axonal targeting of Trk receptors via transcytosis regulates sensitivity to neurotrophin responses. J Neurosci 29: 11674–11685. doi: 10.1523/JNEUROSCI.1542-09.2009
|
| [92] |
Lazo OM, Gonzalez A, Ascano M, et al. (2013) BDNF regulates Rab11-mediated recycling endosome dynamics to induce dendritic branching. J Neurosci 33: 6112–6122. doi: 10.1523/JNEUROSCI.4630-12.2013
|
| [93] |
Chen ZY, Ieraci A, Tanowitz M, et al. (2005) A novel endocytic recycling signal distinguishes biological responses of Trk neurotrophin receptors. Mol Biol Cell 16: 5761–5772. doi: 10.1091/mbc.E05-07-0651
|
| [94] |
Sadowski L, Pilecka I, Miaczynska M (2009) Signaling from endosomes: location makes a difference. Exp Cell Res 315: 1601–1609. doi: 10.1016/j.yexcr.2008.09.021
|
| [95] |
Egea J, Klein R (2007) Bidirectional Eph-ephrin signaling during axon guidance. Trends Cell Biol 17: 230–238. doi: 10.1016/j.tcb.2007.03.004
|
| [96] |
Williams CC, Allison JG, Vidal GA, et al. (2004) The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J Cell Biol 167: 469–478. doi: 10.1083/jcb.200403155
|
| [97] |
Tseng HC, Lyu PC, Lin WC (2010) Nuclear localization of orphan receptor protein kinase (Ror1) is mediated through the juxtamembrane domain. BMC Cell Biol 11: 48. doi: 10.1186/1471-2121-11-48
|
| [98] |
Wang YN, Hung MC (2012) Nuclear functions and subcellular trafficking mechanisms of the epidermal growth factor receptor family. Cell Biosci 2: 13. doi: 10.1186/2045-3701-2-13
|
| [99] |
Schlessinger J, Lemmon MA (2006) Nuclear signaling by receptor tyrosine kinases: the first robin of spring. Cell 127: 45–48. doi: 10.1016/j.cell.2006.09.013
|
| [100] |
Kamio T, Shigematsu K, Sou H, et al. (1990) Immunohistochemical expression of epidermal growth factor receptors in human adrenocortical carcinoma. Hum Pathol 21: 277–282. doi: 10.1016/0046-8177(90)90227-V
|
| [101] |
Marti U, Burwen SJ, Wells A, et al. (1991) Localization of epidermal growth factor receptor in hepatocyte nuclei. Hepatology 13: 15–20. doi: 10.1002/hep.1840130104
|
| [102] |
Lo HW, Ali-Seyed M, Wu Y, et al. (2006) Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J Cell Biochem 98: 1570–1583. doi: 10.1002/jcb.20876
|
| [103] | Brand TM, Iida M, Li C, et al. (2011) The nuclear epidermal growth factor receptor signaling network and its role in cancer. Discov Med 12: 419–432. |
| [104] |
Domingues I, Rino J, Demmers JA, et al. (2011) VEGFR2 translocates to the nucleus to regulate its own transcription. PLoS One 6: e25668. doi: 10.1371/journal.pone.0025668
|
| [105] |
Wang YN, Wang H, Yamaguchi H, et al. (2010) COPI-mediated retrograde trafficking from the Golgi to the ER regulates EGFR nuclear transport. Biochem Biophys Res Commun 399: 498–504. doi: 10.1016/j.bbrc.2010.07.096
|
| [106] |
Forbes SA, Tang G, Bindal N, et al. (2010) COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res 38: D652–657. doi: 10.1093/nar/gkp995
|
| [107] |
Yuzawa S, Opatowsky Y, Zhang Z, et al. (2007) Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 130: 323–334. doi: 10.1016/j.cell.2007.05.055
|
| [108] |
Corless CL, Heinrich MC (2008) Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol 3: 557–586. doi: 10.1146/annurev.pathmechdis.3.121806.151538
|
| [109] |
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674. doi: 10.1016/j.cell.2011.02.013
|
| [110] |
Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285: 1182–1186. doi: 10.1056/NEJM197111182852108
|
| [111] |
Goel HL, Mercurio AM (2013) VEGF targets the tumour cell. Nat Rev Cancer 13: 871–882. doi: 10.1038/nrc3627
|
| [112] |
Lichtenberger BM, Tan PK, Niederleithner H, et al. (2010) Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell 140: 268–279. doi: 10.1016/j.cell.2009.12.046
|
| [113] |
Schoeffner DJ, Matheny SL, Akahane T, et al. (2005) VEGF contributes to mammary tumor growth in transgenic mice through paracrine and autocrine mechanisms. Lab Invest 85: 608–623. doi: 10.1038/labinvest.3700258
|
| [114] |
Ruan GX, Kazlauskas A (2013) Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J Biol Chem 288: 21161–21172. doi: 10.1074/jbc.M113.474619
|
| [115] | Warburg O, Wind F, Negelein E (1927) The Metabolism of Tumors in the Body. J Gen Physiol 8: 519–530. |
| [116] |
Sonveaux P, Copetti T, De Saedeleer CJ, et al. (2012) Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. Plos One 7: e33418. doi: 10.1371/journal.pone.0033418
|
| [117] |
De Saedeleer CJ, Copetti T, Porporato PE, et al. (2012) Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. Plos One 7: e46571. doi: 10.1371/journal.pone.0046571
|
| [118] |
Trabold O, Wagner S, Wicke C, et al. (2003) Lactate and oxygen constitute a fundamental regulatory mechanism in wound healing. Wound Repair Regen 11: 504–509. doi: 10.1046/j.1524-475X.2003.11621.x
|
| [119] |
Porporato PE, Payen VL, De Saedeleer CJ, et al. (2012) Lactate stimulates angiogenesis and accelerates the healing of superficial and ischemic wounds in mice. Angiogenesis 15: 581–592. doi: 10.1007/s10456-012-9282-0
|
| [120] |
Hynes NE, Lane HA (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5: 341–354. doi: 10.1038/nrc1609
|
| [121] | Ohsaki Y, Tanno S, Fujita Y, et al. (2000) Epidermal growth factor receptor expression correlates with poor prognosis in non-small cell lung cancer patients with p53 overexpression. Oncol Rep 7: 603–607. |
| [122] | Nicholson RI, Gee JM, Harper ME (2001) EGFR and cancer prognosis. Eur J Cancer 37 Suppl 4: S9–15. |
| [123] |
Fang K (1996) An enhanced and sensitive autocrine stimulation by transforming growth factor-alpha is acquired in the brain metastatic variant of a human non-small-cell lung cancer cell line. Br J Cancer 74: 1776–1782. doi: 10.1038/bjc.1996.629
|
| [124] | Mendelsohn J (1992) Epidermal growth factor receptor as a target for therapy with antireceptor monoclonal antibodies. J Natl Cancer Inst Monogr 13: 125–131. |
| [125] |
Kosaka T, Yatabe Y, Endoh H, et al. (2004) Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 64: 8919–8923. doi: 10.1158/0008-5472.CAN-04-2818
|
| [126] |
Sharma SV, Bell DW, Settleman J, et al. (2007) Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 7: 169–181. doi: 10.1038/nrc2088
|
| [127] |
Kim KS, Jeong JY, Kim YC, et al. (2005) Predictors of the response to gefitinib in refractory non-small cell lung cancer. Clin Cancer Res 11: 2244–2251. doi: 10.1158/1078-0432.CCR-04-2081
|
| [128] |
Bell DW, Lynch TJ, Haserlat SM, et al. (2005) Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol 23: 8081–8092. doi: 10.1200/JCO.2005.02.7078
|
| [129] |
Jin F, Hagemann N, Brockmeier U, et al. (2013) LDL attenuates VEGF-induced angiogenesis via mechanisms involving VEGFR2 internalization and degradation following endosome-trans-Golgi network trafficking. Angiogenesis 16: 625–637. doi: 10.1007/s10456-013-9340-2
|
| [130] |
Tirziu D, Moodie KL, Zhuang ZW, et al. (2005) Delayed arteriogenesis in hypercholesterolemic mice. Circulation 112: 2501-2509. doi: 10.1161/CIRCULATIONAHA.105.542829
|
| [131] |
Van Belle E, Rivard A, Chen D, et al. (1997) Hypercholesterolemia attenuates angiogenesis but does not preclude augmentation by angiogenic cytokines. Circulation 96: 2667-2674. doi: 10.1161/01.CIR.96.8.2667
|
| [132] |
Rask-Madsen C, King GL (2013) Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metab 17: 20–33. doi: 10.1016/j.cmet.2012.11.012
|
| [133] |
Simons M (2005) Angiogenesis, arteriogenesis, and diabetes: paradigm reassessed? J Am Coll Cardiol 46: 835–837. doi: 10.1016/j.jacc.2005.06.008
|
| [134] |
Werner GS, Richartz BM, Heinke S, et al. (2003) Impaired acute collateral recruitment as a possible mechanism for increased cardiac adverse events in patients with diabetes mellitus. Eur Heart J 24: 1134–1142. doi: 10.1016/S0195-668X(03)00187-8
|
| [135] |
Warren CM, Ziyad S, Briot A, et al. (2014) A ligand-independent VEGFR2 signaling pathway limits angiogenic responses in diabetes. Sci Signal 7: ra1. doi: 10.1126/scisignal.2004235
|
| [136] |
Deng CX, Wynshaw-Boris A, Shen MM, et al. (1994) Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev 8: 3045–3057. doi: 10.1101/gad.8.24.3045
|
| [137] |
Yamaguchi TP, Harpal K, Henkemeyer M, et al. (1994) fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev 8: 3032–3044. doi: 10.1101/gad.8.24.3032
|
| [138] |
White KE, Cabral JM, Davis SI, et al. (2005) Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet 76: 361–367. doi: 10.1086/427956
|
| [139] |
Shiang R, Thompson LM, Zhu Y-Z, et al. (1994) Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 78: 335–342. doi: 10.1016/0092-8674(94)90302-6
|
| [140] |
Jacob AL, Smith C, Partanen J, et al. (2006) Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev Biol 296: 315–328. doi: 10.1016/j.ydbio.2006.05.031
|
| [141] |
Su N, Jin M, Chen L (2014) Role of FGF/FGFR signaling in skeletal development and homeostasis: learning from mouse models. Bone Res 2: 14003. doi: 10.1038/boneres.2014.3
|
| [142] |
Shawver LK, Slamon D, Ullrich A (2002) Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell 1: 117–123. doi: 10.1016/S1535-6108(02)00039-9
|
| [143] |
Ludwig DL, Pereira DS, Zhu Z, et al. (2003) Monoclonal antibody therapeutics and apoptosis. Oncogene 22: 9097–9106. doi: 10.1038/sj.onc.1207104
|
| [144] |
Fauvel B, Yasri A (2014) Antibodies directed against receptor tyrosine kinases: current and future strategies to fight cancer. MAbs 6: 838–851. doi: 10.4161/mabs.29089
|
| [145] |
Ferrara N, Kerbel RS (2005) Angiogenesis as a therapeutic target. Nature 438: 967–974. doi: 10.1038/nature04483
|
| [146] |
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438: 932–936. doi: 10.1038/nature04478
|
| [147] | Fontanella C, Ongaro E, Bolzonello S, et al. (2014) Clinical advances in the development of novel VEGFR2 inhibitors. Ann Transl Med 2: 123. |
| [148] |
Kankanala J, Latham AM, Johnson AP, et al. (2012) A combinatorial in silico and cellular approach to identify a new class of compounds that target VEGFR2 receptor tyrosine kinase activity and angiogenesis. Br J Pharmacol 166: 737–748. doi: 10.1111/j.1476-5381.2011.01801.x
|
| [149] |
Chow LQ, Eckhardt SG (2007) Sunitinib: from rational design to clinical efficacy. J Clin Oncol 25: 884–896. doi: 10.1200/JCO.2006.06.3602
|
| [150] | Ghatalia P, Morgan CJ, Je Y, et al. (2014) Congestive heart failure with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Crit Rev Oncol Hematol. |
| [151] | Jain RK, Duda DG, Clark JW, et al. (2006) Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol 3: 24–40. |
| [152] | Mendel DB, Laird AD, Xin X, et al. (2003) In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 9: 327–337. |
| [153] |
Hasinoff BB, Patel D (2010) The lack of target specificity of small molecule anticancer kinase inhibitors is correlated with their ability to damage myocytes in vitro. Toxicol Appl Pharmacol 249: 132–139. doi: 10.1016/j.taap.2010.08.026
|
| [154] |
Knights V, Cook SJ (2010) De-regulated FGF receptors as therapeutic targets in cancer. Pharmacol Ther 125: 105–117. doi: 10.1016/j.pharmthera.2009.10.001
|
| [155] |
Zhang S, Cao Z, Tian H, et al. (2011) SKLB1002, a novel potent inhibitor of VEGF receptor 2 signaling, inhibits angiogenesis and tumor growth in vivo. Clin Cancer Res 17: 4439–4450. doi: 10.1158/1078-0432.CCR-10-3109
|
| [156] |
Harris PA, Cheung M, Hunter RN, 3rd, et al. (2005) Discovery and evaluation of 2-anilino-5-aryloxazoles as a novel class of VEGFR2 kinase inhibitors. J Med Chem 48: 1610–1619. doi: 10.1021/jm049538w
|
| [157] |
Miyazaki Y, Matsunaga S, Tang J, et al. (2005) Novel 4-amino-furo[2,3-d]pyrimidines as Tie-2 and VEGFR2 dual inhibitors. Bioorg Med Chem Lett 15: 2203–2207. doi: 10.1016/j.bmcl.2005.03.034
|
| [158] |
Latham AM, Bruns AF, Kankanala J, et al. (2012) Indolinones and anilinophthalazines differentially target VEGF-A- and basic fibroblast growth factor-mediated responses in primary human endothelial cells. Br J Pharmacol 165: 245–259. doi: 10.1111/j.1476-5381.2011.01545.x
|
| [159] |
Zhou T, Commodore L, Huang WS, et al. (2010) Structural analysis of DFG-in and DFG-out dual Src-Abl inhibitors sharing a common vinyl purine template. Chem Biol Drug Des 75: 18–28. doi: 10.1111/j.1747-0285.2009.00905.x
|
| [160] |
Imrie H, Abbas A, Viswambharan H, et al. (2009) Vascular insulin-like growth factor-I resistance and diet-induced obesity. Endocrinology 150: 4575–4582. doi: 10.1210/en.2008-1641
|
| [161] | Canfield K, Li J, Wilkins OM, et al. (2015) Receptor tyrosine kinase ERBB4 mediates acquired resistance to ERBB2 inhibitors in breast cancer cells. Cell Cycle 14: 648–655. |
| [162] |
Johnston S, Trudeau M, Kaufman B, et al. (2008) Phase II study of predictive biomarker profiles for response targeting human epidermal growth factor receptor 2 (HER-2) in advanced inflammatory breast cancer with lapatinib monotherapy. J Clin Oncol 26: 1066–1072. doi: 10.1200/JCO.2007.13.9949
|
| [163] | Kaufman B, Stein S, Casey MA, et al. (2008) Lapatinib in combination with capecitabine in the management of ErbB2-positive (HER2-positive) advanced breast cancer. Biologics 2: 61–65. |
| [164] |
Sequist LV, Lynch TJ (2008) EGFR tyrosine kinase inhibitors in lung cancer: an evolving story. Annu Rev Med 59: 429–442. doi: 10.1146/annurev.med.59.090506.202405
|
| [165] | Rusch V, Baselga J, Cordon-Cardo C, et al. (1993) Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res 53: 2379–2385. |
| [166] |
Dutu T, Michiels S, Fouret P, et al. (2005) Differential expression of biomarkers in lung adenocarcinoma: a comparative study between smokers and never-smokers. Ann Oncol 16: 1906–1914. doi: 10.1093/annonc/mdi408
|
| [167] | Wakeling AE, Guy SP, Woodburn JR, et al. (2002) ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res 62: 5749–5754. |
| [168] |
Giaccone G, Herbst RS, Manegold C, et al. (2004) Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 1. J Clin Oncol 22: 777–784. doi: 10.1200/JCO.2004.08.001
|
| [169] |
Herbst RS, Giaccone G, Schiller JH, et al. (2004) Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 2. J Clin Oncol 22: 785–794. doi: 10.1200/JCO.2004.07.215
|
| [170] |
Herbst RS, Prager D, Hermann R, et al. (2005) TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol 23: 5892–5899. doi: 10.1200/JCO.2005.02.840
|
| [171] |
Gatzemeier U, Pluzanska A, Szczesna A, et al. (2007) Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the Tarceva Lung Cancer Investigation Trial. J Clin Oncol 25: 1545–1552. doi: 10.1200/JCO.2005.05.1474
|
| [172] |
Marchetti A, Martella C, Felicioni L, et al. (2005) EGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J Clin Oncol 23: 857–865. doi: 10.1200/JCO.2005.08.043
|
| [173] |
|
| [174] |
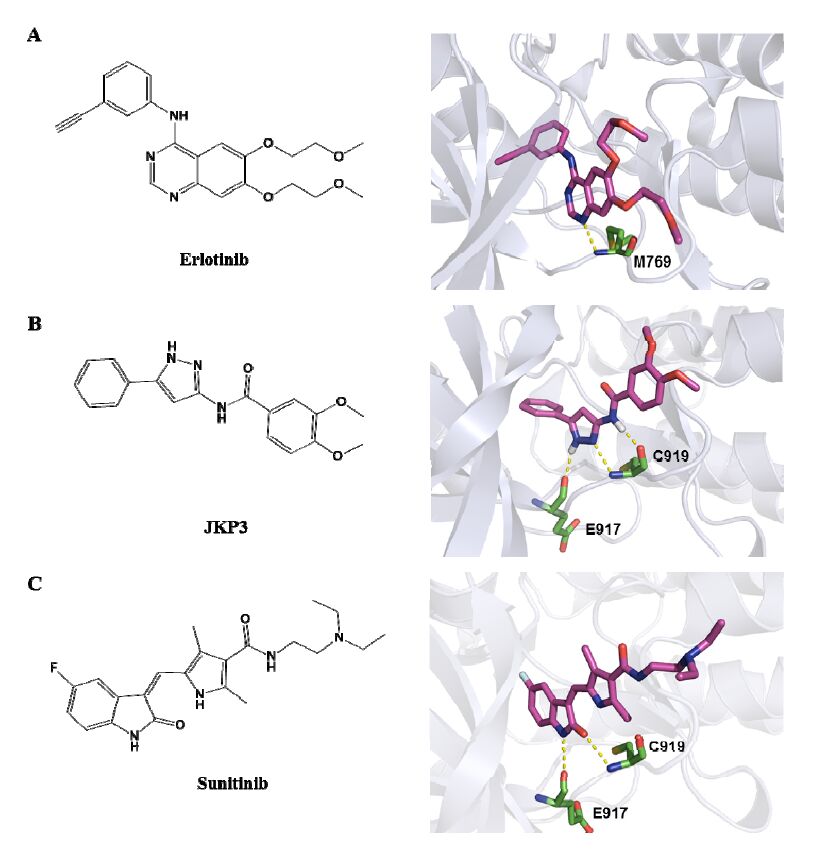
Figures(5) / Tables(1)

Oleg A. Karpov, Gareth W. Fearnley, Gina A. Smith, Jayakanth Kankanala, Michael J. McPherson, Darren C. Tomlinson, Michael A. Harrison, Sreenivasan Ponnambalam. Receptor tyrosine kinase structure and function in health and disease[J]. AIMS Biophysics, 2015, 2(4): 476-502. doi: 10.3934/biophy.2015.4.476







 DownLoad:
DownLoad: