Citation: N. H. AlShamrani, A. M. Elaiw. Stability of an adaptive immunity viral infection model with multi-stages of infected cells and two routes of infection[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 575-605. doi: 10.3934/mbe.2020030

| [1] | M. A. Nowak and C. R. M. Bangham, Population dynamics of immune responses to persistent viruses, Science, 272 (1996), 74-79. |
| [2] | N. Yousfi, K. Hattaf and A. Tridane, Modeling the adaptive immune response in HBV infection, J. Math. Biol., 63 (2011), 933-957. |
| [3] | L. Gibelli, A. Elaiw, M. A. Alghamdi, et al., Heterogeneous population dynamics of active particles: Progression, mutations, and selection dynamics, Math. Models Methods Appl. Sci., 27 (2017), 617-640. |
| [4] | A. M. Elaiw and M. A. Alshaikh, Stability of discrete-time HIV dynamics models with three categories of infected CD4+ T-cells, Adv. Differ. Equ., 2019 (2019), 407. |
| [5] | F. Zhang, J. Li, C. Zheng, et al., Dynamics of an HBV/HCV infection model with intracellular delay and cell proliferation, Commun. Nonlinear Sci. Numer. Simul., 42 (2017), 464-476. |
| [6] | N. Bellomo and Y. Tao, Stabilization in a chemotaxis model for virus infection, Discrete Contin. Dyn. syst. Ser. S, 13 (2020), 105-117. |
| [7] | H. Shu, L. Wang and J. Watmough, Global stability of a nonlinear viral infection model with infinitely distributed intracellular delays and CTL imune responses, SIAM J. Appl. Math., 73 (2013), 1280-1302. |
| [8] | A. M. Elaiw, A. A. Raezah and S. A. Azoz, Stability of delayed HIV dynamics models with two latent reservoirs and immune impairment, Adv. Differ. Equ., 2018 (2018), 414. |
| [9] | M. Y. Li and H. Shu, Global dynamics of a mathematical model for HTLV-I infection of CD4+ T cells with delayed CTL response, Nonlinear Anal. Real World Appl., 13 (2012), 1080-1092. |
| [10] | J. Pang and J-An Cui, Analysis of a hepatitis B viral infection model with immune response delay, Int. J. Biomath., 10 (2017). |
| [11] | C. Vargas-De-Leon, Global properties for a virus dynamics model with lytic and nonlytic immune responses, and nonlinear immune attack rates, J. Biol. Syst., 22 (2014), 449-462. |
| [12] | A. Murase, T. Sasaki and T. Kajiwara, Stability analysis of pathogen-immune interaction dynamics, J. Math. Biol., 51 (2005), 247-267. |
| [13] | A. M. Elaiw and M. A. Alshaikh, Stability analysis of a general discrete-time pathogen infection model with humoral immunity, J. Difference Equ. Appl., 2019 (2019), 1-24. |
| [14] | A. M. Elaiw, E. Kh. Elnahary and A. A. Raezah, Effect of cellular reservoirs and delays on the global dynamics of HIV, Adv. Difference Equ., 2018 (2018), 85. |
| [15] | T. Wang, Z. Hu and F. Liao, Stability and Hopf bifurcation for a virus infection model with delayed humoral immunity response, J. Math. Anal. Appl., 411 (2014) 63-74. |
| [16] | A. M. Elaiw and N. H. AlShamrani, Global stability of humoral immunity virus dynamics models with nonlinear infection rate and removal, Nonlinear Anal. Real World Appl., 26 (2015), 161-190. |
| [17] | H. Miao, Z. Teng, C. Kang, et al., Stability analysis of a virus infection model with humoral immunity response and two time delays, Math. Methods Appl. Sci., 39 (2016), 3434-3449. |
| [18] | D. Wodarz, Killer cell dynamics: Mthematical and computational approaches to immunology, Springer Verlag, New York, 2007. |
| [19] | J. Wang, J. Pang, T. Kuniya, et al., Global threshold dynamics in a five-dimensional virus model with cell-mediated, humoral immune responses and distributed delays, Appl. Math. Comput., 241 (2014), 298-316. |
| [20] | A. Rezounenko, Stability of a viral infection model with state-dependent delay, CTL and antibody immune responses, Discrete Contin. Dyn. Syst.Ser. B, 22 (2017), 1547-156. |
| [21] | Y. Yan and W. Wang, Global stability of a five-dimensional model with immune responses and delay, Discrete Contin. Dyn. Syst.Ser. B, 17 (2012), 401-416. |
| [22] | P. Dubey, U. S. Dubey and B. Dubey, Modeling the role of acquired immune response and antiretroviral therapy in the dynamics of HIV infection, Math. Comput. Simul., 144 (2018), 120- 137. |
| [23] | A. M. Elaiw and N. H. AlShamrani, Stability of an adaptive immunity pathogen dynamics model with latency and multiple delays, Math. Methods Appl. Sci., 36 (2018), 125-142. |
| [24] | R. V. Culshaw, S. Ruan and G. Webb, A mathematical model of cell-to-cell spread of HIV-1 that includes a time delay, J. Math. Biol., 46 (2003), 425-444. |
| [25] | X. Lai and X. Zou, Modeling HIV-1 virus dynamics with both virus-to-cell infection and cell-tocell transmission, SIAM J. Appl. Math., 74 (2014), 898-917. |
| [26] | H. Pourbashash, S. S. Pilyugin, P. De Leenheer, et al., Global analysis of within host virus models with cell-to-cell viral transmission, Discrete Contin. Dyn. Syst.Ser. B, 10 (2014) 3341-3357. |
| [27] | J. Wang, J. Lang and X. Zou, Analysis of an age structured HIV infection model with virus-to-cell infection and cell-to-cell transmission, Nonlinear Anal. Real World Appl., 34 (2017), 75-96. |
| [28] | S. S. Chen, C. Y. Cheng and Y. Takeuchi, Stability analysis in delayed within-host viral dynamics with both viral and cellular infections, J. Math. Anal. Appl., 442 (2016), 642-672. |
| [29] | J. Lin, R. Xu and X. Tian, Threshold dynamics of an HIV-1 virus model with both virus-to-cell and cell-to-cell transmissions, intracellular delay, and humoral immunity, Appl. Math. Comput., 315 (2017), 516-530. |
| [30] | A. M. Elaiw, A. Almatrafi, A. D. Hobiny, et al., Global properties of a general latent pathogen dynamics model with delayed pathogenic and cellular infections, Discrete Dyn. Nat. Soc., 2019 (2019). |
| [31] | A. D. Hobiny, A. M. Elaiw and A. Almatrafi, Stability of delayed pathogen dynamics models with latency and two routes of infection, Adv. Difference Equ., 2018 (2018), 276. |
| [32] | Y. Yang, L. Zou and S. Ruan, Global dynamics of a delayed within-host viral infection model with both virus-to-cell and cell-to-cell transmissions, Math. Biosci., 270 (2015), 183-191. |
| [33] | A. M. Elaiw and A. A. Raezah, Stability of general virus dynamics models with both cellular and viral infections and delays, Math. Methods Appl. Sci., 40 (2017), 5863-5880. |
| [34] | S. Pan and S. P. Chakrabarty, Threshold dynamics of HCV model with cell-to-cell transmission and a non-cytolytic cure in the presence of humoral immunity, Commun. Nonlinear Sci. Numer. Simul., 61 (2018), 180-197. |
| [35] | T. Guo, Z. Qiu and L. Rong, Analysis of an HIV model with immune responses and cell-to-cell transmission, Bull. Malays. Math. Sci. Soc., 2018 (2018), 1-27. |
| [36] | G. Huang, Y. Takeuchi and W. Ma, Lyapunov functionals for delay differential equations model of viral infections, SIAM J. Appl. Math., 70 (2010), 2693-2708. |
| [37] | P. Georgescu and Y. H. Hsieh, Global stability for a virus dynamics model with nonlinear incidence of infection and removal, SIAM J. Appl. Math., 67 (2006), 337-353. |
| [38] | A. M. Elaiw and N. H. AlShamrani, Stability of a general delay-distributed virus dynamics model with multi-staged infected progression and immune response, Math. Methods Appl. Sci., 40 (2017), 699-719. |
| [39] | A. Korobeinikov, Global properties of basic virus dynamics models, Bull. Math. Biol., 66 (2004), 879-883. |
| [40] | A. M. Elaiw and S. A. Azoz, Global properties of a class of HIV infection models with Beddington-DeAngelis functional response, Math. Methods Appl. Sci., 36 (2013), 383-394. |
| [41] | A. M. Elaiw, Global properties of a class of HIV models, Nonlinear Anal. Real World Appl., 11 (2010), 2253-2263. |
| [42] | A. M. Elaiw and N. A. Almuallem, Global properties of delayed-HIV dynamics models with differential drug efficacy in cocirculating target cells, Appl. Math. Comput., 265 (2015), 1067- 1089. |
| [43] | A. M. Elaiw and N. A. Almuallem, Global dynamics of delay-distributed HIV infection models with differential drug efficacy in cocirculating target cells, Math. Methods Appl. Sci., 39 (2016), 4-31. |
| [44] | A. M. Elaiw, I. A. Hassanien and S. A. Azoz, Global stability of HIV infection models with intracellular delays, J. Korean Math. Soc., 49 (2012), 779-794. |
| [45] | A. M. Elaiw, Global properties of a class of virus infection models with multitarget cells, Nonlinear Dynam., 69 (2012), 423-435. |
| [46] | J. K. Hale and S. V. Lunel, Introduction to functional differential equations, Springer-Verlag, New York, 1993. |
| [47] | X. Wang and L. Rong, HIV low viral load persistence under treatment: Insights from a model of cell-to-cell viral transmission, Appl. Math. Lett., 94 (2019), 44-51. |
| [48] | M. Y. Li and H. Shu, Impact of intracellular delays and target-cell dynamics on in vivo viral infections, SIAM J. Appl. Math., 70 (2010), 2434-2448. |
| [49] | A. M. Elaiw and E. Kh. Elnahary, Analysis of general humoral immunity HIV dynamics model with HAART and distributed delays, Mathematics, 7 (2019), 157. |
| [50] | A. M. Elaiw, S. F. Alshehaiween and A. D. Hobiny, Global properties of delay-distributed HIV dynamics model including impairment of B-cell functions, Mathematics, 7 (2019), 837. |
| [51] | C. C. McCluskey, Complete global stability for an SIR epidemic model with delay-distributed or discrete, Nonlinear Anal. Real World Appl., 11 (2010), 55-59. |
| [52] | C. C. McCluskey, Global stability for an SEIR epidemiological model with varying infectivity and infinite delay, Math. Biosci. Eng., 6 (2009), 603-610. |
| [53] | N. Dixit and A. Perelson, Multiplicity of human immunodeficiency virus infections in lymphoid tissue, J. Virol., 78 (2004), 8942-8945. |
| [54] | C. C. McCluskey, Delay versus age-of-infection-Global stability, Appl. Math. Comput., 217 (2010), 3046-3049. |
| [55] | P. Magal, C. C. McCluskey and G. F. Webb, Lyapunov functional and global asymptotic stability for an infection-age model, Appl. Anal., 89 (2010), 1109-1140. |
| [56] | C. Browne, Immune response in virus model structured by cell infection-age, Math. Biosci. Eng., 13 (2016), 887-909. |
| [57] | X. Tian, R. Xu and J. Lin, Mathematical analysis of an age-structured HIV-1 infection model with CTL immune response, Math. Biosci. Eng., 16 (2019), 7850-7882. |
| [58] | A. M. Elaiw and A. D. AlAgha, Global dynamics of reaction-diffusion oncolytic M1 virotherapy with immune response, Appl. Math. Comput., 367 (2020), 124758. |
| [59] | A. Herz, S. Bonhoeffer, R. M. Anderson, et al., Viral dynamics in vivo: Limitations on estimates of intracellular delay and virus decay, Proc.Natl. Acad. Sci., 93 (1996), 7247-7251. |
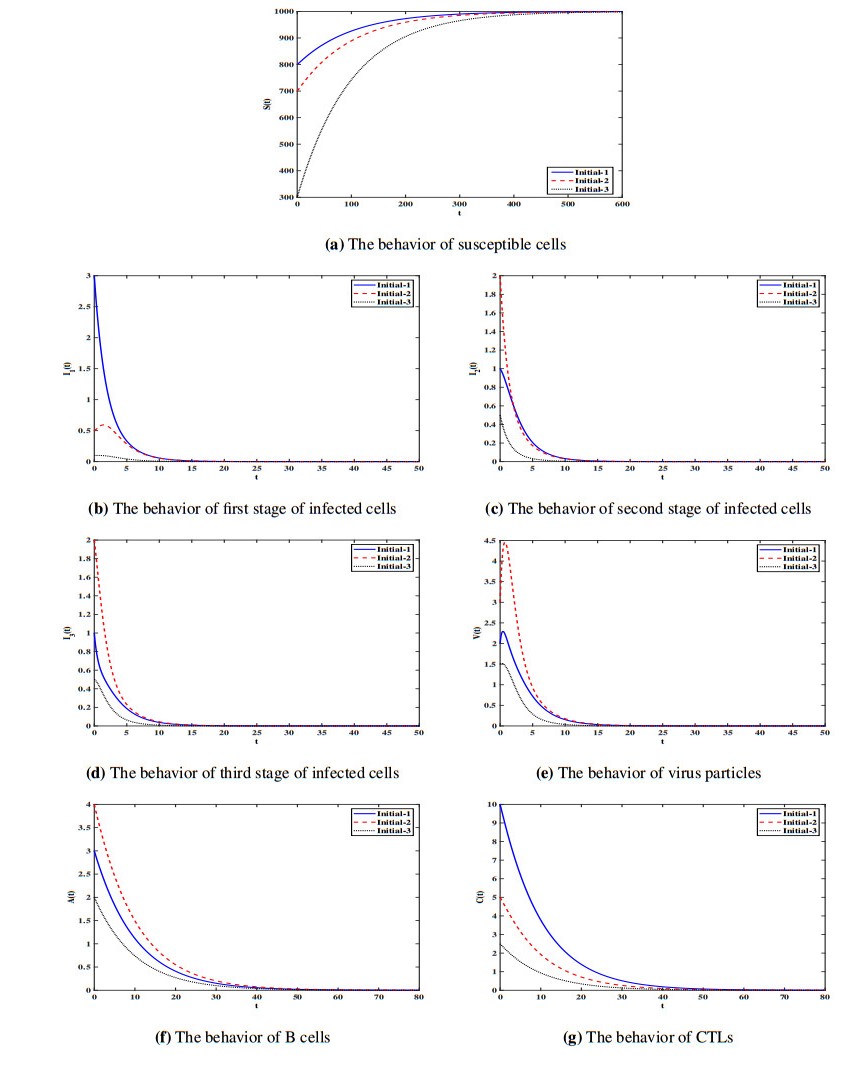
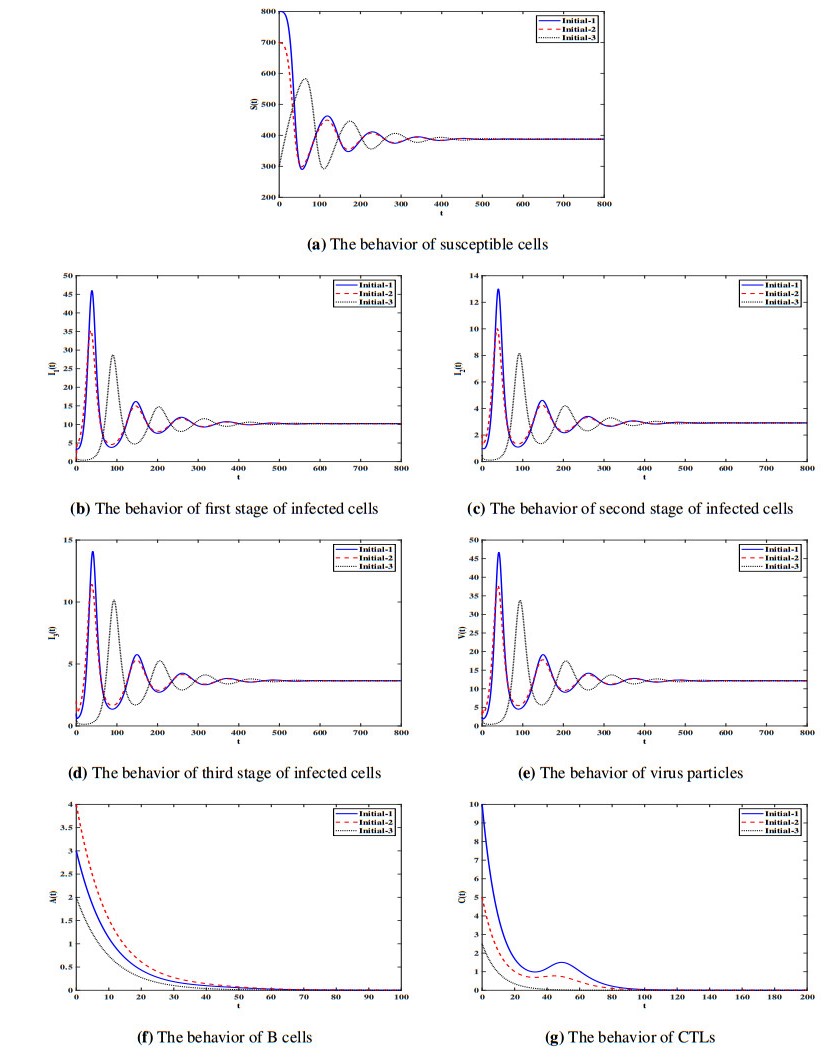
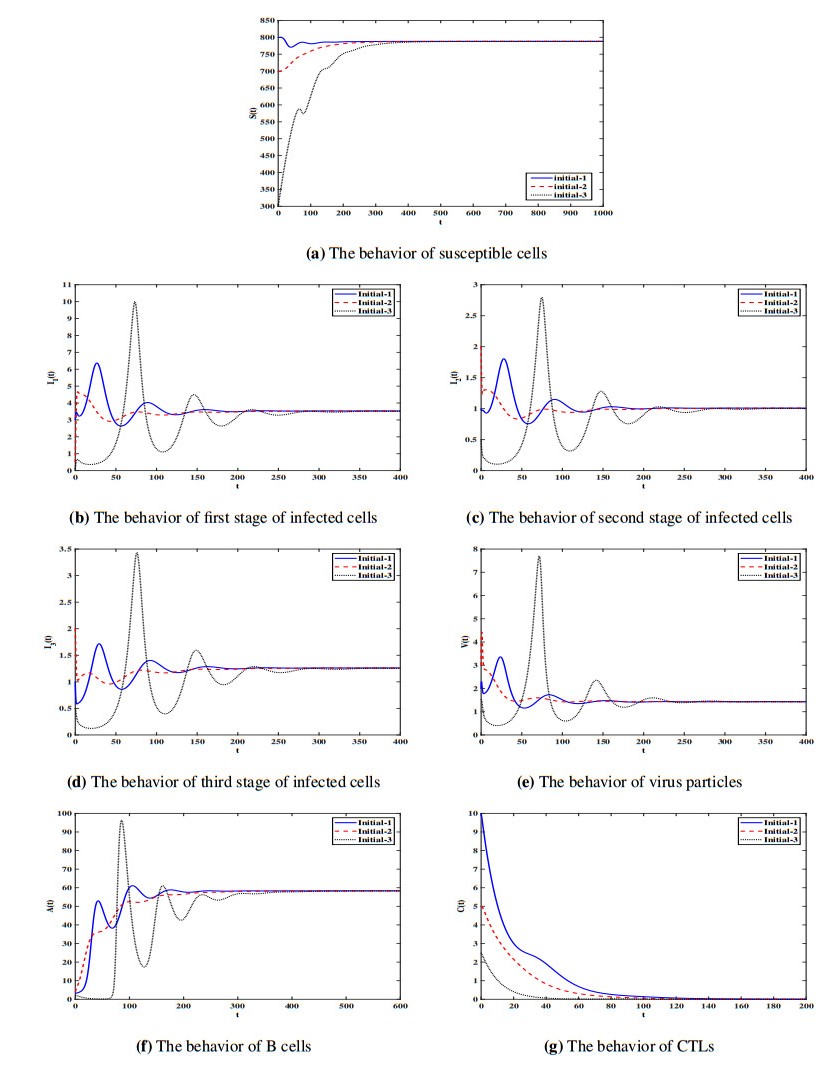
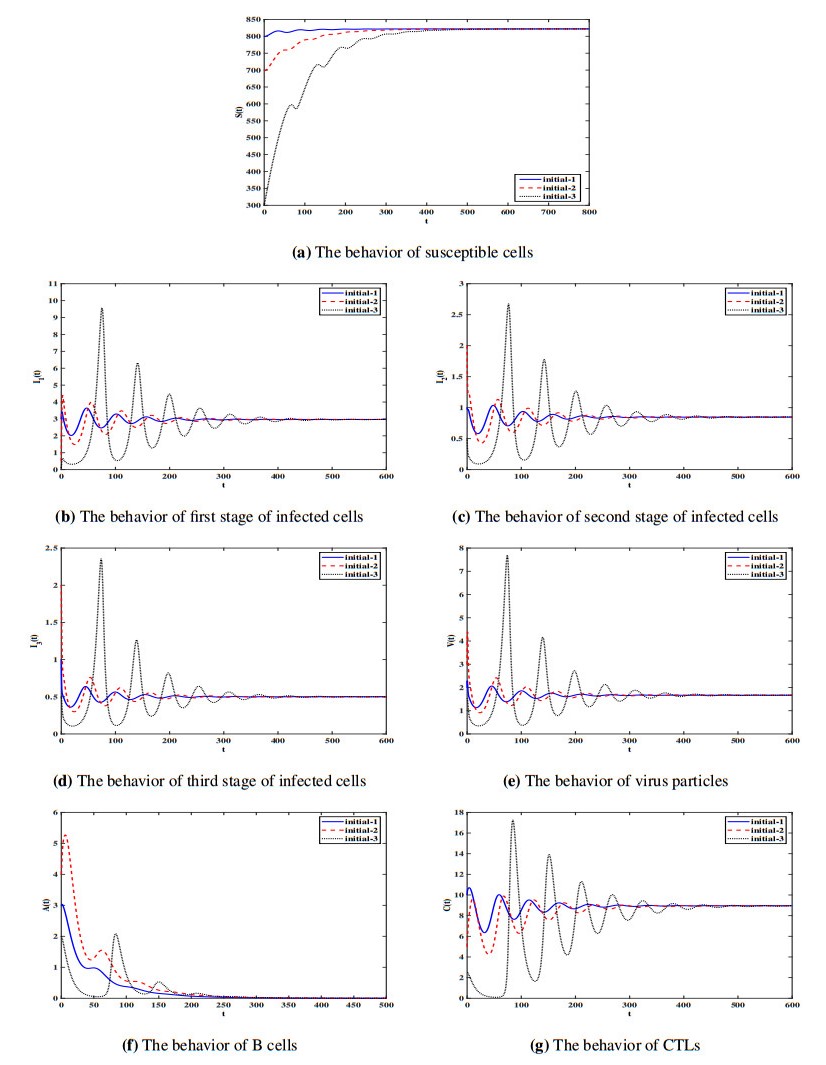
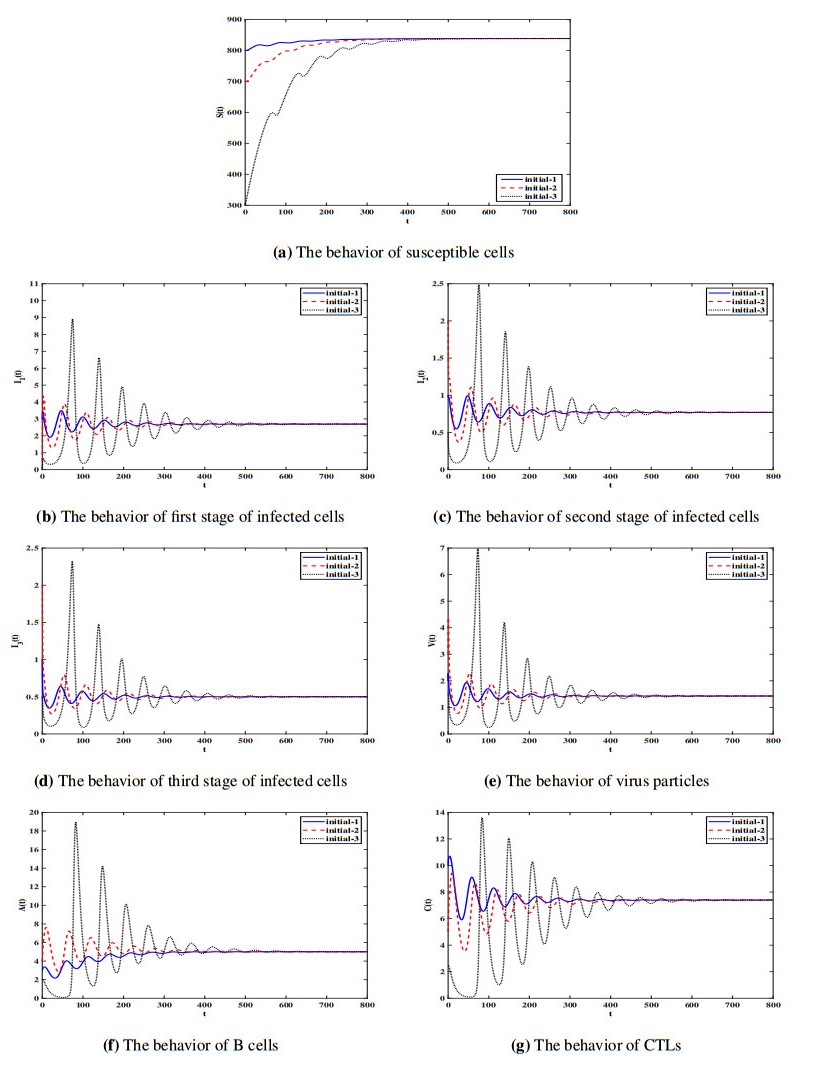
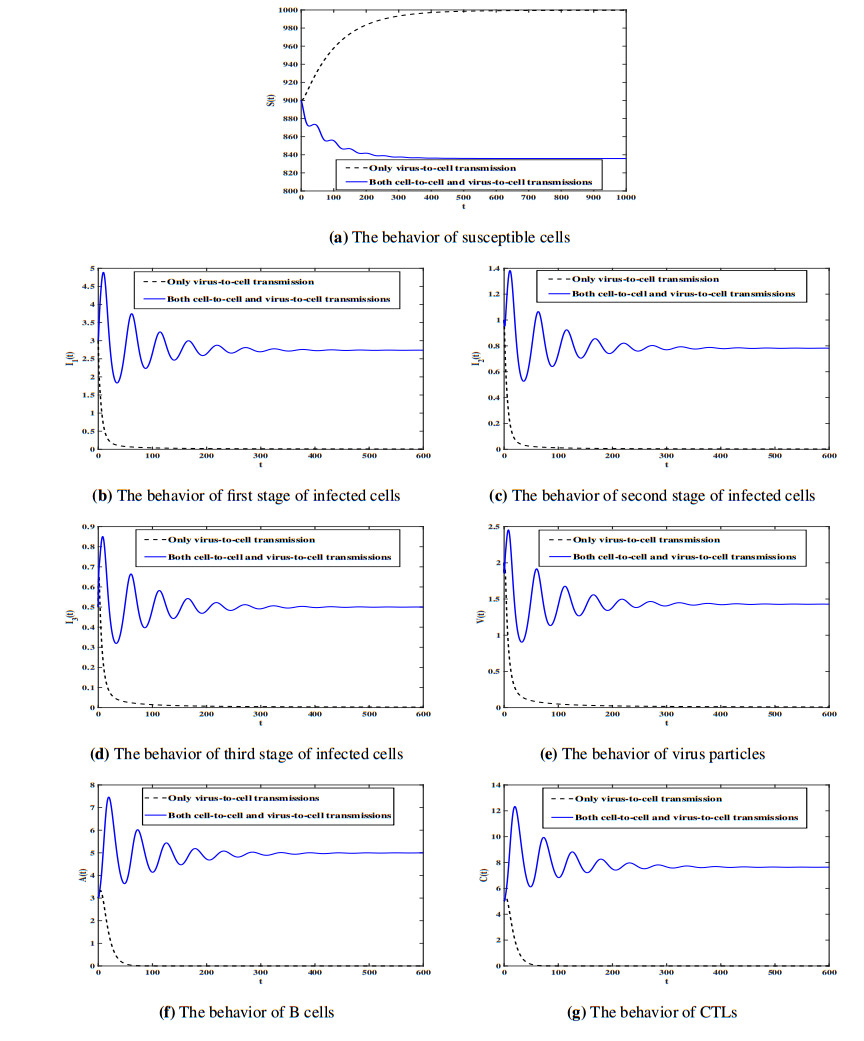
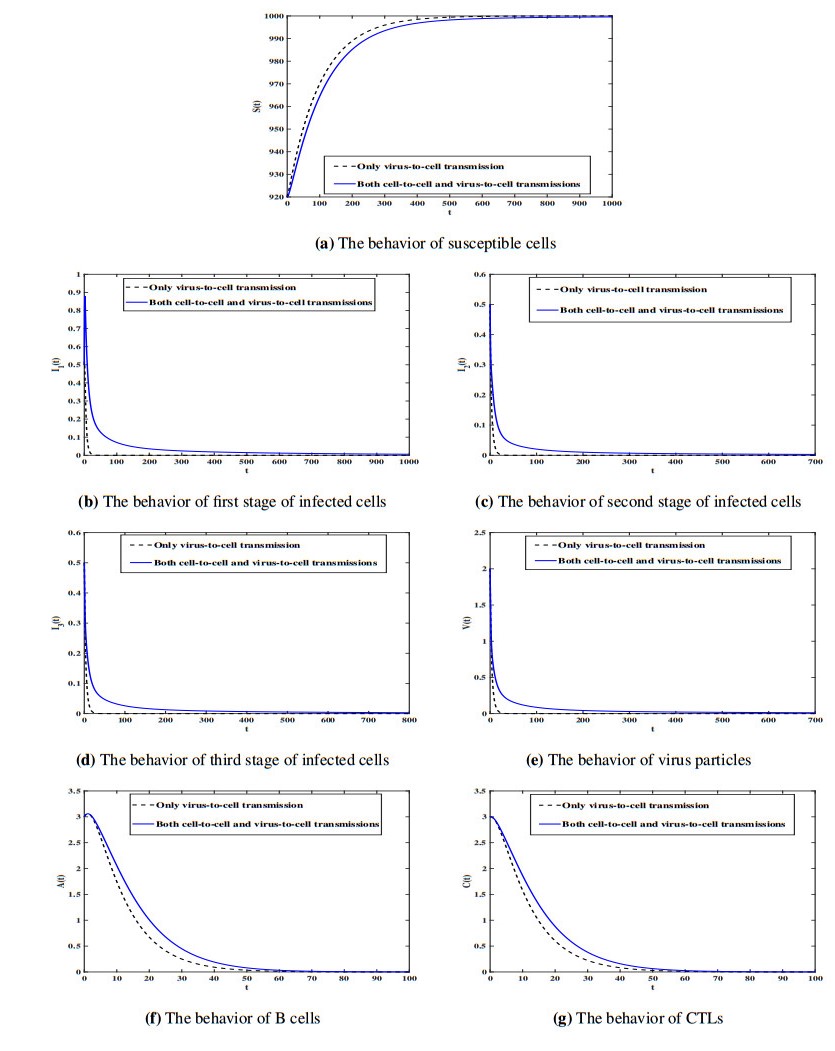
Figures(7) / Tables(1)

N. H. AlShamrani, A. M. Elaiw. Stability of an adaptive immunity viral infection model with multi-stages of infected cells and two routes of infection[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 575-605. doi: 10.3934/mbe.2020030







 DownLoad:
DownLoad: