Citation: Song-Tao Liu, Hang Zhang. The mitotic checkpoint complex (MCC): looking back and forth after 15 years[J]. AIMS Molecular Science, 2016, 3(4): 597-634. doi: 10.3934/molsci.2016.4.597

| [1] |
Hartwell LH, Weinert TA (1989) Checkpoints: controls that ensure the order of cell cycle events. Science 246: 629-634. doi: 10.1126/science.2683079

|
| [2] |
Li R, Murray AW (1991) Feedback control of mitosis in budding yeast. Cell 66: 519-531. doi: 10.1016/0092-8674(81)90015-5
|
| [3] | Hoyt MA, Totis L, Roberts BT (1991) S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 66: 507-517. |
| [4] |
Sudakin V, Ganoth D, Dahan A, et al. (1995) The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell 6: 185-197. doi: 10.1091/mbc.6.2.185
|
| [5] |
King RW, Peters JM, Tugendreich S, et al. (1995) A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 81: 279-288. doi: 10.1016/0092-8674(95)90338-0
|
| [6] |
Irniger S, Piatti S, Michaelis C, et al. (1995) Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell 81: 269-278. doi: 10.1016/0092-8674(95)90337-2
|
| [7] |
Tugendreich S, Tomkiel J, Earnshaw W, et al. (1995) CDC27Hs colocalizes with CDC16Hs to the centrosome and mitotic spindle and is essential for the metaphase to anaphase transition. Cell 81: 261-268. doi: 10.1016/0092-8674(95)90336-4
|
| [8] |
Cohen-Fix O, Peters JM, Kirschner MW, et al. (1996) Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev 10: 3081-3093. doi: 10.1101/gad.10.24.3081
|
| [9] | Funabiki H, Kumada K, Yanagida M (1996) Fission yeast Cut1 and Cut2 are essential for sister chromatid separation, concentrate along the metaphase spindle and form large complexes. EMBO J 15: 6617-6628. |
| [10] |
Hwang LH, Lau LF, Smith DL, et al. (1998) Budding yeast Cdc20: a target of the spindle checkpoint. Science 279: 1041-1044. doi: 10.1126/science.279.5353.1041
|
| [11] |
He X, Patterson TE, Sazer S (1997) The Schizosaccharomyces pombe spindle checkpoint protein mad2p blocks anaphase and genetically interacts with the anaphase-promoting complex. Proc Natl Acad Sci U S A 94: 7965-7970. doi: 10.1073/pnas.94.15.7965
|
| [12] |
Li Y, Gorbea C, Mahaffey D, et al. (1997) MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. Proc Natl Acad Sci U S A 94: 12431-12436. doi: 10.1073/pnas.94.23.12431
|
| [13] |
Fang G, Yu H, Kirschner MW (1998) The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev 12: 1871-1883. doi: 10.1101/gad.12.12.1871
|
| [14] |
Sudakin V, Chan GK, Yen TJ (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol 154: 925-936. doi: 10.1083/jcb.200102093
|
| [15] |
Hardwick KG, Johnston RC, Smith DL, et al. (2000) MAD3 encodes a novel component of the spindle checkpoint which interacts with Bub3p, Cdc20p, and Mad2p. J Cell Biol 148: 871-882. doi: 10.1083/jcb.148.5.871
|
| [16] |
Millband DN, Hardwick KG (2002) Fission yeast Mad3p is required for Mad2p to inhibit the anaphase-promoting complex and localizes to kinetochores in a Bub1p-, Bub3p-, and Mph1p-dependent manner. Mol Cell Biol 22: 2728-2742. doi: 10.1128/MCB.22.8.2728-2742.2002
|
| [17] |
Fraschini R, Beretta A, Sironi L, et al. (2001) Bub3 interaction with Mad2, Mad3 and Cdc20 is mediated by WD40 repeats and does not require intact kinetochores. Embo J 20: 6648-6659. doi: 10.1093/emboj/20.23.6648
|
| [18] |
Chen RH (2002) BubR1 is essential for kinetochore localization of other spindle checkpoint proteins and its phosphorylation requires Mad1. J Cell Biol 158: 487-496. doi: 10.1083/jcb.200204048
|
| [19] |
Chao WC, Kulkarni K, Zhang Z, et al. (2012) Structure of the mitotic checkpoint complex. Nature 484: 208-213. doi: 10.1038/nature10896
|
| [20] |
Alfieri C, Chang L, Zhang Z, et al. (2016) Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 536: 431-436. doi: 10.1038/nature19083
|
| [21] |
Yamaguchi M, VanderLinden R, Weissmann F, et al. (2016) Cryo-EM of Mitotic Checkpoint Complex-Bound APC/C Reveals Reciprocal and Conformational Regulation of Ubiquitin Ligation. Mol Cell 63: 593-607. doi: 10.1016/j.molcel.2016.07.003
|
| [22] |
Sczaniecka M, Feoktistova A, May KM, et al. (2008) The spindle checkpoint functions of Mad3 and Mad2 depend on a Mad3 KEN box-mediated interaction with Cdc20-anaphase-promoting complex (APC/C). J Biol Chem 283: 23039-23047. doi: 10.1074/jbc.M803594200
|
| [23] |
Musacchio A (2015) The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr Biol 25: R1002-1018. doi: 10.1016/j.cub.2015.08.051
|
| [24] | Jia L, Kim S, Yu H (2013) Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem Sci 38 302-311. |
| [25] |
London N, Biggins S (2014) Signalling dynamics in the spindle checkpoint response. Nat Rev Mol Cell Biol 15: 736-747. doi: 10.1038/nrm3888
|
| [26] | Foley EA, Kapoor TM (2013) Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol 14: 25-37. |
| [27] |
Lara-Gonzalez P, Westhorpe FG, Taylor SS (2012) The spindle assembly checkpoint. Curr Biol 22: R966-980. doi: 10.1016/j.cub.2012.10.006
|
| [28] | Stukenberg PT, Burke DJ (2008) The role of the kinetochore in spindle checkpoint signaling. In: De Wulf P, Earnshaw, W.C., eds., editor. The Kinetochore: from Molecular Discoveries to Cancer Therapy. New York: Springer. |
| [29] |
Rieder CL, Cole RW, Khodjakov A, et al. (1995) The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol 130: 941-948. doi: 10.1083/jcb.130.4.941
|
| [30] |
Li X, Nicklas RB (1995) Mitotic forces control a cell-cycle checkpoint. Nature 373: 630-632. doi: 10.1038/373630a0
|
| [31] |
Weiss E, Winey M (1996) The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J Cell Biol 132: 111-123. doi: 10.1083/jcb.132.1.111
|
| [32] |
Suijkerbuijk SJ, van Dam TJ, Karagoz GE, et al. (2012) The vertebrate mitotic checkpoint protein BUBR1 is an unusual pseudokinase. Dev Cell 22: 1321-1329. doi: 10.1016/j.devcel.2012.03.009
|
| [33] |
Vleugel M, Hoogendoorn E, Snel B, et al. (2012) Evolution and function of the mitotic checkpoint. Dev Cell 23: 239-250. doi: 10.1016/j.devcel.2012.06.013
|
| [34] |
Guo Y, Kim C, Ahmad S, et al. (2012) CENP-E--dependent BubR1 autophosphorylation enhances chromosome alignment and the mitotic checkpoint. J Cell Biol 198: 205-217. doi: 10.1083/jcb.201202152
|
| [35] | Musacchio A, Salmon ED (2007) The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8: 379-393. |
| [36] |
Zachos G, Black EJ, Walker M, et al. (2007) Chk1 is required for spindle checkpoint function. Dev Cell 12: 247-260. doi: 10.1016/j.devcel.2007.01.003
|
| [37] |
Chan GK, Jablonski SA, Starr DA, et al. (2000) Human Zw10 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat Cell Biol 2: 944-947. doi: 10.1038/35046598
|
| [38] |
Yao X, Abrieu A, Zheng Y, et al. (2000) CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat Cell Biol 2: 484-491. doi: 10.1038/35019518
|
| [39] |
Williams BC, Li Z, Liu S, et al. (2003) Zwilch, a New Component of the ZW10/ROD Complex Required for Kinetochore Functions. Mol Biol Cell 14: 1379-1391. doi: 10.1091/mbc.E02-09-0624
|
| [40] |
Montembault E, Dutertre S, Prigent C, et al. (2007) PRP4 is a spindle assembly checkpoint protein required for MPS1, MAD1, and MAD2 localization to the kinetochores. J Cell Biol 179: 601-609. doi: 10.1083/jcb.200703133
|
| [41] |
Santaguida S, Vernieri C, Villa F, et al. (2011) Evidence that Aurora B is implicated in spindle checkpoint signalling independently of error correction. Embo J 30: 1508-1519. doi: 10.1038/emboj.2011.70
|
| [42] |
Xia G, Luo X, Habu T, et al. (2004) Conformation-specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. Embo J 23: 3133-3143. doi: 10.1038/sj.emboj.7600322
|
| [43] |
Habu T, Kim SH, Weinstein J, et al. (2002) Identification of a MAD2-binding protein, CMT2, and its role in mitosis. Embo J 21: 6419-6428. doi: 10.1093/emboj/cdf659
|
| [44] |
Tipton AR, Wang K, Oladimeji P, et al. (2012) Identification of novel mitosis regulators through data mining with human centromere/kinetochore proteins as group queries. BMC Cell Biol 13: 15. doi: 10.1186/1471-2121-13-15
|
| [45] |
Wang K, Sturt-Gillespie B, Hittle JC, et al. (2014) Thyroid hormone receptor interacting protein 13 (TRIP13) AAA-ATPase is a novel mitotic checkpoint-silencing protein. J Biol Chem 289: 23928-23937. doi: 10.1074/jbc.M114.585315
|
| [46] |
Eytan E, Wang K, Miniowitz-Shemtov S, et al. (2014) Disassembly of mitotic checkpoint complexes by the joint action of the AAA-ATPase TRIP13 and p31(comet). Proc Natl Acad Sci U S A 111: 12019-12024. doi: 10.1073/pnas.1412901111
|
| [47] |
Gao YF, Li T, Chang Y, et al. (2011) Cdk1-phosphorylated CUEDC2 promotes spindle checkpoint inactivation and chromosomal instability. Nat Cell Biol 13: 924-933. doi: 10.1038/ncb2287
|
| [48] |
Mansfeld J, Collin P, Collins MO, et al. (2011) APC15 drives the turnover of MCC-CDC20 to make the spindle assembly checkpoint responsive to kinetochore attachment. Nat Cell Biol 13: 1234-1243. doi: 10.1038/ncb2347
|
| [49] |
Foster SA, Morgan DO (2012) The APC/C subunit Mnd2/Apc15 promotes Cdc20 autoubiquitination and spindle assembly checkpoint inactivation. Mol Cell 47: 921-932. doi: 10.1016/j.molcel.2012.07.031
|
| [50] |
Uzunova K, Dye BT, Schutz H, et al. (2012) APC15 mediates CDC20 autoubiquitylation by APC/C(MCC) and disassembly of the mitotic checkpoint complex. Nat Struct Mol Biol 19: 1116-1123. doi: 10.1038/nsmb.2412
|
| [51] |
Reddy SK, Rape M, Margansky WA, et al. (2007) Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature 446: 921-925. doi: 10.1038/nature05734
|
| [52] |
Stegmeier F, Rape M, Draviam VM, et al. (2007) Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature 446: 876-881. doi: 10.1038/nature05694
|
| [53] |
Garnett MJ, Mansfeld J, Godwin C, et al. (2009) UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat Cell Biol 11: 1363-1369. doi: 10.1038/ncb1983
|
| [54] |
Griffis ER, Stuurman N, Vale RD (2007) Spindly, a novel protein essential for silencing the spindle assembly checkpoint, recruits dynein to the kinetochore. J Cell Biol 177: 1005-1015. doi: 10.1083/jcb.200702062
|
| [55] |
Liu D, Vleugel M, Backer CB, et al. (2010) Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J Cell Biol 188: 809-820. doi: 10.1083/jcb.201001006
|
| [56] |
Rosenberg JS, Cross FR, Funabiki H (2011) KNL1/Spc105 recruits PP1 to silence the spindle assembly checkpoint. Curr Biol 21: 942-947. doi: 10.1016/j.cub.2011.04.011
|
| [57] |
Howell BJ, McEwen BF, Canman JC, et al. (2001) Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol 155: 1159-1172. doi: 10.1083/jcb.200105093
|
| [58] |
Foley EA, Maldonado M, Kapoor TM (2011) Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat Cell Biol 13: 1265-1271. doi: 10.1038/ncb2327
|
| [59] |
Suijkerbuijk SJ, Vleugel M, Teixeira A, et al. (2012) Integration of kinase and phosphatase activities by BUBR1 ensures formation of stable kinetochore-microtubule attachments. Dev Cell 23: 745-755. doi: 10.1016/j.devcel.2012.09.005
|
| [60] |
Pinsky BA, Biggins S (2005) The spindle checkpoint: tension versus attachment. Trends Cell Biol 15: 486-493. doi: 10.1016/j.tcb.2005.07.005
|
| [61] | King JM, Nicklas RB (2000) Tension on chromosomes increases the number of kinetochore microtubules but only within limits. J Cell Sci 113 Pt 21: 3815-3823. |
| [62] |
Wilson L, Jordan MA (1995) Microtubule dynamics: taking aim at a moving target. Chem Biol 2: 569-573. doi: 10.1016/1074-5521(95)90119-1
|
| [63] |
Wilson L, Panda D, Jordan MA (1999) Modulation of microtubule dynamics by drugs: a paradigm for the actions of cellular regulators. Cell Struct Funct 24: 329-335. doi: 10.1247/csf.24.329
|
| [64] |
Skoufias DA, Andreassen PR, Lacroix FB, et al. (2001) Mammalian mad2 and bub1/bubR1 recognize distinct spindle-attachment and kinetochore-tension checkpoints. Proc Natl Acad Sci U S A 98: 4492-4497. doi: 10.1073/pnas.081076898
|
| [65] |
Kapoor TM, Mayer TU, Coughlin ML, et al. (2000) Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol 150: 975-988. doi: 10.1083/jcb.150.5.975
|
| [66] |
Collin P, Nashchekina O, Walker R, et al. (2013) The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat Cell Biol 15: 1378-1385. doi: 10.1038/ncb2855
|
| [67] |
Dick AE, Gerlich DW (2013) Kinetic framework of spindle assembly checkpoint signalling. Nat Cell Biol 15: 1370-1377. doi: 10.1038/ncb2842
|
| [68] |
Chen RH, Waters JC, Salmon ED, et al. (1996) Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science 274: 242-246. doi: 10.1126/science.274.5285.242
|
| [69] |
Li Y, Benezra R (1996) Identification of a human mitotic checkpoint gene: hsMAD2. Science 274: 246-248. doi: 10.1126/science.274.5285.246
|
| [70] |
Mao Y, Abrieu A, Cleveland DW (2003) Activating and silencing the mitotic checkpoint through CENP-E-dependent activation/inactivation of BubR1. Cell 114: 87-98. doi: 10.1016/S0092-8674(03)00475-6
|
| [71] | Ji Z, Gao H, Yu H (2015) CELL DIVISION CYCLE. Kinetochore attachment sensed by competitive Mps1 and microtubule binding to Ndc80C. Science 348: 1260-1264. |
| [72] | Hiruma Y, Sacristan C, Pachis ST, et al. (2015) CELL DIVISION CYCLE. Competition between MPS1 and microtubules at kinetochores regulates spindle checkpoint signaling. Science 348: 1264-1267. |
| [73] |
Aravamudhan P, Goldfarb AA, Joglekar AP (2015) The kinetochore encodes a mechanical switch to disrupt spindle assembly checkpoint signalling. Nat Cell Biol 17: 868-879. doi: 10.1038/ncb3179
|
| [74] |
Luo X, Yu H (2008) Protein metamorphosis: the two-state behavior of Mad2. Structure 16: 1616-1625. doi: 10.1016/j.str.2008.10.002
|
| [75] |
Mapelli M, Musacchio A (2007) MAD contortions: conformational dimerization boosts spindle checkpoint signaling. Curr Opin Struct Biol 17: 716-725. doi: 10.1016/j.sbi.2007.08.011
|
| [76] | Peters JM (2006) The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 7: 644-656. |
| [77] |
Barford D (2011) Structural insights into anaphase-promoting complex function and mechanism. Philos Trans R Soc Lond B Biol Sci 366: 3605-3624. doi: 10.1098/rstb.2011.0069
|
| [78] | Chang L, Barford D (2014) Insights into the anaphase-promoting complex: a molecular machine that regulates mitosis. Curr Opin Struct Biol 29: 1-9. |
| [79] |
Qiao R, Weissmann F, Yamaguchi M, et al. (2016) Mechanism of APC/CCDC20 activation by mitotic phosphorylation. Proc Natl Acad Sci U S A 113: E2570-2578. doi: 10.1073/pnas.1604929113
|
| [80] |
Zhang S, Chang L, Alfieri C, et al. (2016) Molecular mechanism of APC/C activation by mitotic phosphorylation. Nature 533: 260-264. doi: 10.1038/nature17973
|
| [81] |
Steen JA, Steen H, Georgi A, et al. (2008) Different phosphorylation states of the anaphase promoting complex in response to antimitotic drugs: a quantitative proteomic analysis. Proc Natl Acad Sci U S A 105: 6069-6074. doi: 10.1073/pnas.0709807104
|
| [82] |
Fujimitsu K, Grimaldi M, Yamano H (2016) Cyclin-dependent kinase 1-dependent activation of APC/C ubiquitin ligase. Science 352: 1121-1124. doi: 10.1126/science.aad3925
|
| [83] |
Kimata Y, Baxter JE, Fry AM, et al. (2008) A role for the Fizzy/Cdc20 family of proteins in activation of the APC/C distinct from substrate recruitment. Mol Cell 32: 576-583. doi: 10.1016/j.molcel.2008.09.023
|
| [84] |
Glotzer M, Murray AW, Kirschner MW (1991) Cyclin is degraded by the ubiquitin pathway. Nature 349: 132-138. doi: 10.1038/349132a0
|
| [85] | Pfleger CM, Kirschner MW (2000) The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev 14: 655-665. |
| [86] |
Di Fiore B, Davey NE, Hagting A, et al. (2015) The ABBA motif binds APC/C activators and is shared by APC/C substrates and regulators. Dev Cell 32: 358-372. doi: 10.1016/j.devcel.2015.01.003
|
| [87] |
Reis A, Levasseur M, Chang HY, et al. (2006) The CRY box: a second APCcdh1-dependent degron in mammalian cdc20. EMBO Rep 7: 1040-1045. doi: 10.1038/sj.embor.7400772
|
| [88] |
Burton JL, Xiong Y, Solomon MJ (2011) Mechanisms of pseudosubstrate inhibition of the anaphase promoting complex by Acm1. EMBO J 30: 1818-1829. doi: 10.1038/emboj.2011.90
|
| [89] |
Diaz-Martinez LA, Tian W, Li B, et al. (2015) The Cdc20-binding Phe box of the spindle checkpoint protein BubR1 maintains the mitotic checkpoint complex during mitosis. J Biol Chem 290: 2431-2443. doi: 10.1074/jbc.M114.616490
|
| [90] |
Lischetti T, Zhang G, Sedgwick GG, et al. (2014) The internal Cdc20 binding site in BubR1 facilitates both spindle assembly checkpoint signalling and silencing. Nat Commun 5: 5563. doi: 10.1038/ncomms6563
|
| [91] |
Elowe S, Dulla K, Uldschmid A, et al. (2010) Uncoupling of the spindle-checkpoint and chromosome-congression functions of BubR1. J Cell Sci 123: 84-94. doi: 10.1242/jcs.056507
|
| [92] |
Morgan DO (2016) Cell division: Mitotic regulation comes into focus. Nature 536: 407-408. doi: 10.1038/nature19423
|
| [93] |
Tipton AR, Wang K, Link L, et al. (2011) BUBR1 and Closed MAD2 (C-MAD2) Interact Directly to Assemble a Functional Mitotic Checkpoint Complex. J Biol Chem 286: 21173-21179. doi: 10.1074/jbc.M111.238543
|
| [94] | Izawa D, Pines J (2015) The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature 517: 631-634. |
| [95] |
Lara-Gonzalez P, Scott MI, Diez M, et al. (2011) BubR1 blocks substrate recruitment to the APC/C in a KEN-box-dependent manner. J Cell Sci 124: 4332-4345. doi: 10.1242/jcs.094763
|
| [96] |
Larsen NA, Al-Bassam J, Wei RR, et al. (2007) Structural analysis of Bub3 interactions in the mitotic spindle checkpoint. Proc Natl Acad Sci U S A 104: 1201-1206. doi: 10.1073/pnas.0610358104
|
| [97] |
Larsen NA, Harrison SC (2004) Crystal structure of the spindle assembly checkpoint protein Bub3. J Mol Biol 344: 885-892. doi: 10.1016/j.jmb.2004.09.094
|
| [98] | Primorac I, Weir JR, Chiroli E, et al. (2013) Bub3 reads phosphorylated MELT repeats to promote spindle assembly checkpoint signaling. Elife 2: e01030. |
| [99] | Overlack K, Primorac I, Vleugel M, et al. (2015) A molecular basis for the differential roles of Bub1 and BubR1 in the spindle assembly checkpoint. Elife 4: e05269. |
| [100] |
Taylor SS, Ha E, McKeon F (1998) The human homologue of Bub3 is required for kinetochore localization of Bub1 and a Mad3/Bub1-related protein kinase. J Cell Biol 142: 1-11. doi: 10.1083/jcb.142.1.1
|
| [101] |
Yu H (2007) Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell 27: 3-16. doi: 10.1016/j.molcel.2007.06.009
|
| [102] |
Visintin R, Prinz S, Amon A (1997) CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science 278: 460-463. doi: 10.1126/science.278.5337.460
|
| [103] |
Schwab M, Lutum AS, Seufert W (1997) Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 90: 683-693. doi: 10.1016/S0092-8674(00)80529-2
|
| [104] |
Tian W, Li B, Warrington R, et al. (2012) Structural analysis of human Cdc20 supports multisite degron recognition by APC/C. Proc Natl Acad Sci U S A 109: 18419-18424. doi: 10.1073/pnas.1213438109
|
| [105] |
He J, Chao WC, Zhang Z, et al. (2013) Insights into degron recognition by APC/C coactivators from the structure of an Acm1-Cdh1 complex. Mol Cell 50: 649-660. doi: 10.1016/j.molcel.2013.04.024
|
| [106] |
Chang L, Zhang Z, Yang J, et al. (2014) Molecular architecture and mechanism of the anaphase-promoting complex. Nature 513: 388-393. doi: 10.1038/nature13543
|
| [107] |
Buschhorn BA, Petzold G, Galova M, et al. (2011) Substrate binding on the APC/C occurs between the coactivator Cdh1 and the processivity factor Doc1. Nat Struct Mol Biol 18: 6-13. doi: 10.1038/nsmb.1979
|
| [108] |
da Fonseca PC, Kong EH, Zhang Z, et al. (2011) Structures of APC/C(Cdh1) with substrates identify Cdh1 and Apc10 as the D-box co-receptor. Nature 470: 274-278. doi: 10.1038/nature09625
|
| [109] |
Izawa D, Pines J (2011) How APC/C-Cdc20 changes its substrate specificity in mitosis. Nat Cell Biol 13: 223-233. doi: 10.1038/ncb2165
|
| [110] |
Schwab M, Neutzner M, Mocker D, et al. (2001) Yeast Hct1 recognizes the mitotic cyclin Clb2 and other substrates of the ubiquitin ligase APC. Embo J 20: 5165-5175. doi: 10.1093/emboj/20.18.5165
|
| [111] |
Vodermaier HC, Gieffers C, Maurer-Stroh S, et al. (2003) TPR subunits of the anaphase-promoting complex mediate binding to the activator protein CDH1. Curr Biol 13: 1459-1468. doi: 10.1016/S0960-9822(03)00581-5
|
| [112] |
Luo X, Tang Z, Rizo J, et al. (2002) The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell 9: 59-71. doi: 10.1016/S1097-2765(01)00435-X
|
| [113] |
Izawa D, Pines J (2012) Mad2 and the APC/C compete for the same site on Cdc20 to ensure proper chromosome segregation. J Cell Biol 199: 27-37. doi: 10.1083/jcb.201205170
|
| [114] |
Mondal G, Baral RN, Roychoudhury S (2006) A new Mad2-interacting domain of Cdc20 is critical for the function of Mad2-Cdc20 complex in the spindle assembly checkpoint. Biochem J 396: 243-253. doi: 10.1042/BJ20051914
|
| [115] | Aravind L, Koonin EV (1998) The HORMA domain: a common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem Sci 23: 284-286. |
| [116] |
Luo X, Tang Z, Xia G, et al. (2004) The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol 11: 338-345. doi: 10.1038/nsmb748
|
| [117] |
Sironi L, Mapelli M, Knapp S, et al. (2002) Crystal structure of the tetrameric Mad1-Mad2 core complex: implications of a 'safety belt' binding mechanism for the spindle checkpoint. Embo J 21: 2496-2506. doi: 10.1093/emboj/21.10.2496
|
| [118] |
Sironi L, Melixetian M, Faretta M, et al. (2001) Mad2 binding to Mad1 and Cdc20, rather than oligomerization, is required for the spindle checkpoint. Embo J 20: 6371-6382. doi: 10.1093/emboj/20.22.6371
|
| [119] |
Luo X, Fang G, Coldiron M, et al. (2000) Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. Nat Struct Biol 7: 224-229. doi: 10.1038/73338
|
| [120] |
Orth M, Mayer B, Rehm K, et al. (2011) Shugoshin is a Mad1/Cdc20-like interactor of Mad2. Embo J 30: 2868-2880. doi: 10.1038/emboj.2011.187
|
| [121] |
Lee SH, McCormick F, Saya H (2010) Mad2 inhibits the mitotic kinesin MKlp2. J Cell Biol 191: 1069-1077. doi: 10.1083/jcb.201003095
|
| [122] | Schibler A, Koutelou E, Tomida J, et al. (2016) Histone H3K4 methylation regulates deactivation of the spindle assembly checkpoint through direct binding of Mad2. Genes Dev 30: 1187-1197. |
| [123] |
Mapelli M, Massimiliano L, Santaguida S, et al. (2007) The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell 131: 730-743. doi: 10.1016/j.cell.2007.08.049
|
| [124] |
Yang M, Li B, Tomchick DR, et al. (2007) p31comet blocks Mad2 activation through structural mimicry. Cell 131: 744-755. doi: 10.1016/j.cell.2007.08.048
|
| [125] |
Yang M, Li B, Liu CJ, et al. (2008) Insights into mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric mad2 dimer. PLoS Biol 6: e50. doi: 10.1371/journal.pbio.0060050
|
| [126] |
Skinner JJ, Wood S, Shorter J, et al. (2008) The Mad2 partial unfolding model: regulating mitosis through Mad2 conformational switching. J Cell Biol 183: 761-768. doi: 10.1083/jcb.200808122
|
| [127] |
De Antoni A, Pearson CG, Cimini D, et al. (2005) The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol 15: 214-225. doi: 10.1016/j.cub.2005.01.038
|
| [128] | Campbell MS, Chan GK, Yen TJ (2001) Mitotic checkpoint proteins HsMAD1 and HsMAD2 are associated with nuclear pore complexes in interphase. J Cell Sci 114: 953-963. |
| [129] |
Chen RH, Brady DM, Smith D, et al. (1999) The spindle checkpoint of budding yeast depends on a tight complex between the Mad1 and Mad2 proteins. Mol Biol Cell 10: 2607-2618. doi: 10.1091/mbc.10.8.2607
|
| [130] |
Martin-Lluesma S, Stucke VM, Nigg EA (2002) Role of hec1 in spindle checkpoint signaling and kinetochore recruitment of mad1/mad2. Science 297: 2267-2270. doi: 10.1126/science.1075596
|
| [131] |
Hewitt L, Tighe A, Santaguida S, et al. (2010) Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J Cell Biol 190: 25-34. doi: 10.1083/jcb.201002133
|
| [132] |
Tipton AR, Ji W, Sturt-Gillespie B, et al. (2013) Monopolar Spindle 1 (MPS1) Kinase Promotes Production of Closed MAD2 (C-MAD2) Conformer and Assembly of the Mitotic Checkpoint Complex. J Biol Chem 288: 35149-35158. doi: 10.1074/jbc.M113.522375
|
| [133] |
Liu ST, Chan GK, Hittle JC, et al. (2003) Human MPS1 Kinase Is Required for Mitotic Arrest Induced by the Loss of CENP-E from Kinetochores. Mol Biol Cell 14: 1638-1651. doi: 10.1091/mbc.02-05-0074
|
| [134] |
Liu ST, Hittle JC, Jablonski SA, et al. (2003) Human CENP-I specifies localization of CENP-F, MAD1 and MAD2 to kinetochores and is essential for mitosis. Nat Cell Biol 5: 341-345. doi: 10.1038/ncb953
|
| [135] |
Liu ST, Rattner JB, Jablonski SA, et al. (2006) Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J Cell Biol 175: 41-53. doi: 10.1083/jcb.200606020
|
| [136] |
Tipton AR, Tipton M, Yen T, et al. (2011) Closed MAD2 (C-MAD2) is selectively incorporated into the mitotic checkpoint complex (MCC). Cell Cycle 10: 3740-3750. doi: 10.4161/cc.10.21.17919
|
| [137] |
Tang Z, Bharadwaj R, Li B, et al. (2001) Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev Cell 1: 227-237. doi: 10.1016/S1534-5807(01)00019-3
|
| [138] |
Fang G (2002) Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol Biol Cell 13: 755-766. doi: 10.1091/mbc.01-09-0437
|
| [139] |
Kulukian A, Han JS, Cleveland DW (2009) Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev Cell 16: 105-117. doi: 10.1016/j.devcel.2008.11.005
|
| [140] |
Han JS, Holland AJ, Fachinetti D, et al. (2013) Catalytic Assembly of the Mitotic Checkpoint Inhibitor BubR1-Cdc20 by a Mad2-Induced Functional Switch in Cdc20. Mol Cell 51: 92-104. doi: 10.1016/j.molcel.2013.05.019
|
| [141] |
Westhorpe FG, Tighe A, Lara-Gonzalez P, et al. (2011) p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J Cell Sci 124: 3905-3916. doi: 10.1242/jcs.093286
|
| [142] |
Miller JJ, Summers MK, Hansen DV, et al. (2006) Emi1 stably binds and inhibits the anaphase-promoting complex/cyclosome as a pseudosubstrate inhibitor. Genes Dev 20: 2410-2420. doi: 10.1101/gad.1454006
|
| [143] |
Eytan E, Braunstein I, Ganoth D, et al. (2008) Two different mitotic checkpoint inhibitors of the anaphase-promoting complex/cyclosome antagonize the action of the activator Cdc20. Proc Natl Acad Sci U S A 105: 9181-9185. doi: 10.1073/pnas.0804069105
|
| [144] |
Poddar A, Stukenberg PT, Burke DJ (2005) Two complexes of spindle checkpoint proteins containing Cdc20 and Mad2 assemble during mitosis independently of the kinetochore in Saccharomyces cerevisiae. Eukaryot Cell 4: 867-878. doi: 10.1128/EC.4.5.867-878.2005
|
| [145] | Weinstein J (1997) Cell cycle-regulated expression, phosphorylation, and degradation of p55Cdc. A mammalian homolog of CDC20/Fizzy/slp1. J Biol Chem 272: 28501-28511. |
| [146] |
Wolthuis R, Clay-Farrace L, van Zon W, et al. (2008) Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol Cell 30: 290-302. doi: 10.1016/j.molcel.2008.02.027
|
| [147] |
Malureanu LA, Jeganathan KB, Hamada M, et al. (2009) BubR1 N terminus acts as a soluble inhibitor of cyclin B degradation by APC/C(Cdc20) in interphase. Dev Cell 16: 118-131. doi: 10.1016/j.devcel.2008.11.004
|
| [148] |
Ma HT, Poon RY (2011) Orderly inactivation of the key checkpoint protein mitotic arrest deficient 2 (MAD2) during mitotic progression. J Biol Chem 286: 13052-13059. doi: 10.1074/jbc.M110.201897
|
| [149] |
Michel LS, Liberal V, Chatterjee A, et al. (2001) MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 409: 355-359. doi: 10.1038/35053094
|
| [150] |
Hernando E, Nahle Z, Juan G, et al. (2004) Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 430: 797-802. doi: 10.1038/nature02820
|
| [151] |
Schvartzman JM, Duijf PH, Sotillo R, et al. (2011) Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 19: 701-714. doi: 10.1016/j.ccr.2011.04.017
|
| [152] |
Herzog F, Primorac I, Dube P, et al. (2009) Structure of the anaphase-promoting complex/cyclosome interacting with a mitotic checkpoint complex. Science 323: 1477-1481. doi: 10.1126/science.1163300
|
| [153] |
Kramer ER, Scheuringer N, Podtelejnikov AV, et al. (2000) Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol Biol Cell 11: 1555-1569. doi: 10.1091/mbc.11.5.1555
|
| [154] | Simonetta M, Manzoni R, Mosca R, et al. (2009) The influence of catalysis on mad2 activation dynamics. PLoS Biol 7: e10. |
| [155] |
Nilsson J, Yekezare M, Minshull J, et al. (2008) The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat Cell Biol 10: 1411-1420. doi: 10.1038/ncb1799
|
| [156] |
Kallio M, Weinstein J, Daum JR, et al. (1998) Mammalian p55CDC mediates association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase-promoting complex, and is involved in regulating anaphase onset and late mitotic events. J Cell Biol 141: 1393-1406. doi: 10.1083/jcb.141.6.1393
|
| [157] |
Diaz-Martinez LA, Yu H (2007) Running on a treadmill: dynamic inhibition of APC/C by the spindle checkpoint. Cell Div 2: 23. doi: 10.1186/1747-1028-2-23
|
| [158] |
Fava LL, Kaulich M, Nigg EA, et al. (2011) Probing the in vivo function of Mad1:C-Mad2 in the spindle assembly checkpoint. Embo J 30: 3322-3336. doi: 10.1038/emboj.2011.239
|
| [159] |
Sedgwick GG, Larsen MS, Lischetti T, et al. (2016) Conformation-specific anti-Mad2 monoclonal antibodies for the dissection of checkpoint signaling. MAbs 8: 689-697. doi: 10.1080/19420862.2016.1160988
|
| [160] |
Davenport J, Harris LD, Goorha R (2006) Spindle checkpoint function requires Mad2-dependent Cdc20 binding to the Mad3 homology domain of BubR1. Exp Cell Res 312: 1831-1842. doi: 10.1016/j.yexcr.2006.02.018
|
| [161] |
Murray AW, Kirschner MW (1989) Dominoes and clocks: the union of two views of the cell cycle. Science 246: 614-621. doi: 10.1126/science.2683077
|
| [162] |
Meraldi P, Draviam VM, Sorger PK (2004) Timing and checkpoints in the regulation of mitotic progression. Dev Cell 7: 45-60. doi: 10.1016/j.devcel.2004.06.006
|
| [163] |
Waters JC, Chen RH, Murray AW, et al. (1998) Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J Cell Biol 141: 1181-1191. doi: 10.1083/jcb.141.5.1181
|
| [164] |
Hauf S, Cole RW, LaTerra S, et al. (2003) The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol 161: 281-294. doi: 10.1083/jcb.200208092
|
| [165] |
Canman JC, Sharma N, Straight A, et al. (2002) Anaphase onset does not require the microtubule-dependent depletion of kinetochore and centromere-binding proteins. J Cell Sci 115: 3787-3795. doi: 10.1242/jcs.00057
|
| [166] |
Ma HT, Chan YY, Chen X, et al. (2012) Depletion of p31comet protein promotes sensitivity to antimitotic drugs. J Biol Chem 287: 21561-21569. doi: 10.1074/jbc.M112.364356
|
| [167] |
Brown NG, VanderLinden R, Watson ER, et al. (2016) Dual RING E3 Architectures Regulate Multiubiquitination and Ubiquitin Chain Elongation by APC/C. Cell 165: 1440-1453. doi: 10.1016/j.cell.2016.05.037
|
| [168] |
Brown NG, Watson ER, Weissmann F, et al. (2014) Mechanism of polyubiquitination by human anaphase-promoting complex: RING repurposing for ubiquitin chain assembly. Mol Cell 56: 246-260. doi: 10.1016/j.molcel.2014.09.009
|
| [169] |
Primorac I, Musacchio A (2013) Panta rhei: the APC/C at steady state. J Cell Biol 201: 177-189. doi: 10.1083/jcb.201301130
|
| [170] |
Jin L, Williamson A, Banerjee S, et al. (2008) Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell 133: 653-665. doi: 10.1016/j.cell.2008.04.012
|
| [171] |
Williamson A, Wickliffe KE, Mellone BG, et al. (2009) Identification of a physiological E2 module for the human anaphase-promoting complex. Proc Natl Acad Sci U S A 106: 18213-18218. doi: 10.1073/pnas.0907887106
|
| [172] |
Dimova NV, Hathaway NA, Lee BH, et al. (2012) APC/C-mediated multiple monoubiquitylation provides an alternative degradation signal for cyclin B1. Nat Cell Biol 14: 168-176. doi: 10.1038/ncb2425
|
| [173] |
Meyer HJ, Rape M (2014) Enhanced protein degradation by branched ubiquitin chains. Cell 157: 910-921. doi: 10.1016/j.cell.2014.03.037
|
| [174] |
Tang Z, Li B, Bharadwaj R, et al. (2001) APC2 Cullin protein and APC11 RING protein comprise the minimal ubiquitin ligase module of the anaphase-promoting complex. Mol Biol Cell 12: 3839-3851. doi: 10.1091/mbc.12.12.3839
|
| [175] |
Brown NG, VanderLinden R, Watson ER, et al. (2015) RING E3 mechanism for ubiquitin ligation to a disordered substrate visualized for human anaphase-promoting complex. Proc Natl Acad Sci U S A 112: 5272-5279. doi: 10.1073/pnas.1504161112
|
| [176] |
Fang G, Yu H, Kirschner MW (1998) Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol Cell 2: 163-171. doi: 10.1016/S1097-2765(00)80126-4
|
| [177] |
Chang L, Zhang Z, Yang J, et al. (2015) Atomic structure of the APC/C and its mechanism of protein ubiquitination. Nature 522: 450-454. doi: 10.1038/nature14471
|
| [178] |
Kelly A, Wickliffe KE, Song L, et al. (2014) Ubiquitin chain elongation requires E3-dependent tracking of the emerging conjugate. Mol Cell 56: 232-245. doi: 10.1016/j.molcel.2014.09.010
|
| [179] |
Braunstein I, Miniowitz S, Moshe Y, et al. (2007) Inhibitory factors associated with anaphase-promoting complex/cylosome in mitotic checkpoint. Proc Natl Acad Sci U S A 104: 4870-4875. doi: 10.1073/pnas.0700523104
|
| [180] |
Miniowitz-Shemtov S, Teichner A, Sitry-Shevah D, et al. (2010) ATP is required for the release of the anaphase-promoting complex/cyclosome from inhibition by the mitotic checkpoint. Proc Natl Acad Sci U S A 107: 5351-5356. doi: 10.1073/pnas.1001875107
|
| [181] |
Pan J, Chen RH (2004) Spindle checkpoint regulates Cdc20p stability in Saccharomyces cerevisiae. Genes Dev 18: 1439-1451. doi: 10.1101/gad.1184204
|
| [182] |
Foe IT, Foster SA, Cheung SK, et al. (2011) Ubiquitination of Cdc20 by the APC occurs through an intramolecular mechanism. Curr Biol 21: 1870-1877. doi: 10.1016/j.cub.2011.09.051
|
| [183] |
Ge S, Skaar JR, Pagano M (2009) APC/C- and Mad2-mediated degradation of Cdc20 during spindle checkpoint activation. Cell Cycle 8: 167-171. doi: 10.4161/cc.8.1.7606
|
| [184] |
Jia L, Li B, Warrington RT, et al. (2011) Defining pathways of spindle checkpoint silencing: functional redundancy between Cdc20 ubiquitination and p31(comet). Mol Biol Cell 22: 4227-4235. doi: 10.1091/mbc.E11-05-0389
|
| [185] |
Varetti G, Guida C, Santaguida S, et al. (2011) Homeostatic control of mitotic arrest. Mol Cell 44: 710-720. doi: 10.1016/j.molcel.2011.11.014
|
| [186] |
King EM, van der Sar SJ, Hardwick KG (2007) Mad3 KEN boxes mediate both Cdc20 and Mad3 turnover, and are critical for the spindle checkpoint. PLoS ONE 2: e342. doi: 10.1371/journal.pone.0000342
|
| [187] |
Zhang Y, Foreman O, Wigle DA, et al. (2012) USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J Clin Invest 122: 4362-4374. doi: 10.1172/JCI63084
|
| [188] |
Zhang Y, van Deursen J, Galardy PJ (2011) Overexpression of ubiquitin specific protease 44 (USP44) induces chromosomal instability and is frequently observed in human T-cell leukemia. PLoS One 6: e23389. doi: 10.1371/journal.pone.0023389
|
| [189] |
Kim S, Yu H (2011) Mutual regulation between the spindle checkpoint and APC/C. Semin Cell Dev Biol 22: 551-558. doi: 10.1016/j.semcdb.2011.03.008
|
| [190] |
Zeng X, Sigoillot F, Gaur S, et al. (2010) Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 18: 382-395. doi: 10.1016/j.ccr.2010.08.010
|
| [191] |
Bezler A, Gonczy P (2010) Mutual antagonism between the anaphase promoting complex and the spindle assembly checkpoint contributes to mitotic timing in Caenorhabditis elegans. Genetics 186: 1271-1283. doi: 10.1534/genetics.110.123133
|
| [192] |
Chesnel F, Bazile F, Pascal A, et al. (2006) Cyclin B dissociation from CDK1 precedes its degradation upon MPF inactivation in mitotic extracts of Xenopus laevis embryos. Cell Cycle 5: 1687-1698. doi: 10.4161/cc.5.15.3123
|
| [193] |
Holt LJ, Krutchinsky AN, Morgan DO (2008) Positive feedback sharpens the anaphase switch. Nature 454: 353-357. doi: 10.1038/nature07050
|
| [194] | Ye Q, Rosenberg SC, Moeller A, et al. (2015) TRIP13 is a protein-remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. Elife 4. |
| [195] |
Vader G (2015) Pch2(TRIP13): controlling cell division through regulation of HORMA domains. Chromosoma 124: 333-339. doi: 10.1007/s00412-015-0516-y
|
| [196] | Musacchio A (2015) Closing the Mad2 cycle. Elife 4. |
| [197] |
Verdugo A, Vinod PK, Tyson JJ, et al. (2013) Molecular mechanisms creating bistable switches at cell cycle transitions. Open Biol 3: 120179. doi: 10.1098/rsob.120179
|
| [198] |
Hauf S (2013) The spindle assembly checkpoint: progress and persistent puzzles. Biochem Soc Trans 41: 1755-1760. doi: 10.1042/BST20130240
|
| [199] |
Hagan RS, Manak MS, Buch HK, et al. (2011) p31(comet) acts to ensure timely spindle checkpoint silencing subsequent to kinetochore attachment. Mol Biol Cell 22: 4236-4246. doi: 10.1091/mbc.E11-03-0216
|
| [200] |
Teichner A, Eytan E, Sitry-Shevah D, et al. (2011) p31comet promotes disassembly of the mitotic checkpoint complex in an ATP-dependent process. Proc Natl Acad Sci U S A 108: 3187-3192. doi: 10.1073/pnas.1100023108
|
| [201] |
Miniowitz-Shemtov S, Eytan E, Ganoth D, et al. (2012) Role of phosphorylation of Cdc20 in p31(comet)-stimulated disassembly of the mitotic checkpoint complex. Proc Natl Acad Sci U S A 109: 8056-8060. doi: 10.1073/pnas.1204081109
|
| [202] |
Carter SL, Eklund AC, Kohane IS, et al. (2006) A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet 38: 1043-1048. doi: 10.1038/ng1861
|
| [203] |
Martin KJ, Patrick DR, Bissell MJ, et al. (2008) Prognostic breast cancer signature identified from 3D culture model accurately predicts clinical outcome across independent datasets. PLoS ONE 3: e2994. doi: 10.1371/journal.pone.0002994
|
| [204] |
Rhodes DR, Yu J, Shanker K, et al. (2004) Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A 101: 9309-9314. doi: 10.1073/pnas.0401994101
|
| [205] | Neuwald AF, Aravind L, Spouge JL, et al. (1999) AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9: 27-43. |
| [206] |
Ma HT, Poon RY (2016) TRIP13 Regulates Both the Activation and Inactivation of the Spindle-Assembly Checkpoint. Cell Rep 14: 1086-1099. doi: 10.1016/j.celrep.2016.01.001
|
| [207] |
Nelson CR, Hwang T, Chen PH, et al. (2015) TRIP13PCH-2 promotes Mad2 localization to unattached kinetochores in the spindle checkpoint response. J Cell Biol 211: 503-516. doi: 10.1083/jcb.201505114
|
| [208] | Hardwick KG, Shah JV (2010) Spindle checkpoint silencing: ensuring rapid and concerted anaphase onset. F1000 Biol Rep 2: 55. |
| [209] |
Vanoosthuyse V, Hardwick KG (2009) Overcoming inhibition in the spindle checkpoint. Genes Dev 23: 2799-2805. doi: 10.1101/gad.1882109
|
| [210] |
Chan YW, Fava LL, Uldschmid A, et al. (2009) Mitotic control of kinetochore-associated dynein and spindle orientation by human Spindly. J Cell Biol 185: 859-874. doi: 10.1083/jcb.200812167
|
| [211] |
Gassmann R, Holland AJ, Varma D, et al. (2010) Removal of Spindly from microtubule-attached kinetochores controls spindle checkpoint silencing in human cells. Genes Dev 24: 957-971. doi: 10.1101/gad.1886810
|
| [212] |
Meadows JC, Shepperd LA, Vanoosthuyse V, et al. (2011) Spindle checkpoint silencing requires association of PP1 to both Spc7 and kinesin-8 motors. Dev Cell 20: 739-750. doi: 10.1016/j.devcel.2011.05.008
|
| [213] | Porter IM, Schleicher K, Porter M, et al. (2013) Bod1 regulates protein phosphatase 2A at mitotic kinetochores. Nat Commun 4: 2677. |
| [214] |
Pinsky BA, Kotwaliwale CV, Tatsutani SY, et al. (2006) Glc7/protein phosphatase 1 regulatory subunits can oppose the Ipl1/aurora protein kinase by redistributing Glc7. Mol Cell Biol 26: 2648-2660. doi: 10.1128/MCB.26.7.2648-2660.2006
|
| [215] |
Vanoosthuyse V, Meadows JC, van der Sar SJ, et al. (2009) Bub3p facilitates spindle checkpoint silencing in fission yeast. Mol Biol Cell 20: 5096-5105. doi: 10.1091/mbc.E09-09-0762
|
| [216] |
Kim T, Moyle MW, Lara-Gonzalez P, et al. (2015) Kinetochore-localized BUB-1/BUB-3 complex promotes anaphase onset in C. elegans. J Cell Biol 209: 507-517. doi: 10.1083/jcb.201412035
|
| [217] |
Yang Y, Lacefield S (2016) Bub3 activation and inhibition of the APC/C. Cell Cycle 15: 1-2. doi: 10.1080/15384101.2015.1106746
|
| [218] |
Windecker H, Langegger M, Heinrich S, et al. (2009) Bub1 and Bub3 promote the conversion from monopolar to bipolar chromosome attachment independently of shugoshin. EMBO Rep 10: 1022-1028. doi: 10.1038/embor.2009.183
|
| [219] |
Yang Y, Tsuchiya D, Lacefield S (2015) Bub3 promotes Cdc20-dependent activation of the APC/C in S. cerevisiae. J Cell Biol 209: 519-527. doi: 10.1083/jcb.201412036
|
| [220] |
Han JS, Vitre B, Fachinetti D, et al. (2014) Bimodal activation of BubR1 by Bub3 sustains mitotic checkpoint signaling. Proc Natl Acad Sci U S A 111: E4185-4193. doi: 10.1073/pnas.1416277111
|
| [221] |
Kim S, Sun H, Ball HL, et al. (2010) Phosphorylation of the spindle checkpoint protein Mad2 regulates its conformational transition. Proc Natl Acad Sci U S A 107: 19772-19777. doi: 10.1073/pnas.1009000107
|
| [222] |
Wassmann K, Liberal V, Benezra R (2003) Mad2 phosphorylation regulates its association with Mad1 and the APC/C. Embo J 22: 797-806. doi: 10.1093/emboj/cdg071
|
| [223] |
Elowe S, Hummer S, Uldschmid A, et al. (2007) Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev 21: 2205-2219. doi: 10.1101/gad.436007
|
| [224] |
Matsumura S, Toyoshima F, Nishida E (2007) Polo-like kinase 1 facilitates chromosome alignment during prometaphase through BubR1. J Biol Chem 282: 15217-15227. doi: 10.1074/jbc.M611053200
|
| [225] |
Chan GK, Jablonski SA, Sudakin V, et al. (1999) Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J Cell Biol 146: 941-954. doi: 10.1083/jcb.146.5.941
|
| [226] |
Dou Z, von Schubert C, Korner R, et al. (2011) Quantitative mass spectrometry analysis reveals similar substrate consensus motif for human Mps1 kinase and Plk1. PLoS ONE 6: e18793. doi: 10.1371/journal.pone.0018793
|
| [227] |
Huang H, Hittle J, Zappacosta F, et al. (2008) Phosphorylation sites in BubR1 that regulate kinetochore attachment, tension, and mitotic exit. J Cell Biol 183: 667-680. doi: 10.1083/jcb.200805163
|
| [228] |
Mao Y, Desai A, Cleveland DW (2005) Microtubule capture by CENP-E silences BubR1-dependent mitotic checkpoint signaling. J Cell Biol 170: 873-880. doi: 10.1083/jcb.200505040
|
| [229] |
Elowe S (2011) Bub1 and BubR1: at the interface between chromosome attachment and the spindle checkpoint. Mol Cell Biol 31: 3085-3093. doi: 10.1128/MCB.05326-11
|
| [230] |
Bolanos-Garcia VM, Blundell TL (2011) BUB1 and BUBR1: multifaceted kinases of the cell cycle. Trends Biochem Sci 36: 141-150. doi: 10.1016/j.tibs.2010.08.004
|
| [231] |
Chung E, Chen RH (2003) Phosphorylation of Cdc20 is required for its inhibition by the spindle checkpoint. Nat Cell Biol 5: 748-753. doi: 10.1038/ncb1022
|
| [232] |
Kallio M, Mustalahti T, Yen TJ, et al. (1998) Immunolocalization of alpha-tubulin, gamma-tubulin, and CENP-E in male rat and male mouse meiotic divisions: pathway of meiosis I spindle formation in mammalian spermatocytes. Dev Biol 195: 29-37. doi: 10.1006/dbio.1997.8822
|
| [233] |
Wu H, Lan Z, Li W, et al. (2000) p55CDC/hCDC20 is associated with BUBR1 and may be a downstream target of the spindle checkpoint kinase. Oncogene 19: 4557-4562. doi: 10.1038/sj.onc.1203803
|
| [234] | Zich J, Hardwick KG (2009) Getting down to the phosphorylated 'nuts and bolts' of spindle checkpoint signalling. Trends Biochem Sci. |
| [235] |
Hornbeck PV, Zhang B, Murray B, et al. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43: D512-520. doi: 10.1093/nar/gku1267
|
| [236] |
Kim M, Murphy K, Liu F, et al. (2005) Caspase-mediated specific cleavage of BubR1 is a determinant of mitotic progression. Mol Cell Biol 25: 9232-9248. doi: 10.1128/MCB.25.21.9232-9248.2005
|
| [237] | Baek KH, Shin HJ, Jeong SJ, et al. (2005) Caspases-dependent cleavage of mitotic checkpoint proteins in response to microtubule inhibitor. Oncol Res 15: 161-168. |
| [238] |
Choi E, Choe H, Min J, et al. (2009) BubR1 acetylation at prometaphase is required for modulating APC/C activity and timing of mitosis. Embo J 28: 2077-2089. doi: 10.1038/emboj.2009.123
|
| [239] |
Park I, Lee HO, Choi E, et al. (2013) Loss of BubR1 acetylation causes defects in spindle assembly checkpoint signaling and promotes tumor formation. J Cell Biol 202: 295-309. doi: 10.1083/jcb.201210099
|
| [240] |
Yang F, Hu L, Chen C, et al. (2012) BubR1 is modified by sumoylation during mitotic progression. J Biol Chem 287: 4875-4882. doi: 10.1074/jbc.M111.318261
|
| [241] |
Yang F, Huang Y, Dai W (2012) Sumoylated BubR1 plays an important role in chromosome segregation and mitotic timing. Cell Cycle 11: 797-806. doi: 10.4161/cc.11.4.19307
|
| [242] |
Doncic A, Ben-Jacob E, Barkai N (2005) Evaluating putative mechanisms of the mitotic spindle checkpoint. Proc Natl Acad Sci U S A 102: 6332-6337. doi: 10.1073/pnas.0409142102
|
| [243] |
Sear RP, Howard M (2006) Modeling dual pathways for the metazoan spindle assembly checkpoint. Proc Natl Acad Sci U S A 103: 16758-16763. doi: 10.1073/pnas.0603174103
|
| [244] |
Mistry HB, MacCallum DE, Jackson RC, et al. (2008) Modeling the temporal evolution of the spindle assembly checkpoint and role of Aurora B kinase. Proc Natl Acad Sci U S A 105: 20215-20220. doi: 10.1073/pnas.0810706106
|
| [245] |
Chen J, Liu J (2014) Spatial-temporal model for silencing of the mitotic spindle assembly checkpoint. Nat Commun 5: 4795. doi: 10.1038/ncomms5795
|
| [246] |
Chen J, Liu J (2016) Spindle Size Scaling Contributes to Robust Silencing of Mitotic Spindle Assembly Checkpoint. Biophys J 111: 1064-1077. doi: 10.1016/j.bpj.2016.07.039
|
| [247] |
Ibrahim B (2015) Toward a systems-level view of mitotic checkpoints. Prog Biophys Mol Biol 117: 217-224. doi: 10.1016/j.pbiomolbio.2015.02.005
|
| [248] |
Ibrahim B (2015) Spindle assembly checkpoint is sufficient for complete Cdc20 sequestering in mitotic control. Comput Struct Biotechnol J 13: 320-328. doi: 10.1016/j.csbj.2015.03.006
|
| [249] |
Ibrahim B, Dittrich P, Diekmann S, et al. (2008) Mad2 binding is not sufficient for complete Cdc20 sequestering in mitotic transition control (an in silico study). Biophys Chem 134: 93-100. doi: 10.1016/j.bpc.2008.01.007
|
| [250] |
Ibrahim B, Schmitt E, Dittrich P, et al. (2009) In silico study of kinetochore control, amplification, and inhibition effects in MCC assembly. Biosystems 95: 35-50. doi: 10.1016/j.biosystems.2008.06.007
|
| [251] |
Ciliberto A, Shah JV (2009) A quantitative systems view of the spindle assembly checkpoint. Embo J 28: 2162-2173. doi: 10.1038/emboj.2009.186
|
| [252] |
Burton JL, Solomon MJ (2007) Mad3p, a pseudosubstrate inhibitor of APCCdc20 in the spindle assembly checkpoint. Genes Dev 21: 655-667. doi: 10.1101/gad.1511107
|
| [253] |
D'Arcy S, Davies OR, Blundell TL, et al. (2010) Defining the molecular basis of BubR1 kinetochore interactions and APC/C-CDC20 inhibition. J Biol Chem 285: 14764-14776. doi: 10.1074/jbc.M109.082016
|
| [254] |
Bolanos-Garcia VM, Lischetti T, Matak-Vinkovic D, et al. (2011) Structure of a Blinkin-BUBR1 complex reveals an interaction crucial for kinetochore-mitotic checkpoint regulation via an unanticipated binding Site. Structure 19: 1691-1700. doi: 10.1016/j.str.2011.09.017
|
| [255] |
Krenn V, Wehenkel A, Li X, et al. (2012) Structural analysis reveals features of the spindle checkpoint kinase Bub1-kinetochore subunit Knl1 interaction. J Cell Biol 196: 451-467. doi: 10.1083/jcb.201110013
|
| [256] |
Harris L, Davenport J, Neale G, et al. (2005) The mitotic checkpoint gene BubR1 has two distinct functions in mitosis. Exp Cell Res 308: 85-100. doi: 10.1016/j.yexcr.2005.03.036
|
| [257] |
Zhang Y, Lees E (2001) Identification of an overlapping binding domain on Cdc20 for Mad2 and anaphase-promoting complex: model for spindle checkpoint regulation. Mol Cell Biol 21: 5190-5199. doi: 10.1128/MCB.21.15.5190-5199.2001
|
| [258] |
Sethi N, Monteagudo MC, Koshland D, et al. (1991) The CDC20 gene product of Saccharomyces cerevisiae, a beta-transducin homolog, is required for a subset of microtubule-dependent cellular processes. Mol Cell Biol 11: 5592-5602. doi: 10.1128/MCB.11.11.5592
|
| [259] |
Rosenberg SC, Corbett KD (2015) The multifaceted roles of the HORMA domain in cellular signaling. J Cell Biol 211: 745-755. doi: 10.1083/jcb.201509076
|
| [260] |
Mapelli M, Filipp FV, Rancati G, et al. (2006) Determinants of conformational dimerization of Mad2 and its inhibition by p31comet. Embo J 25: 1273-1284 doi: 10.1038/sj.emboj.7601033
|
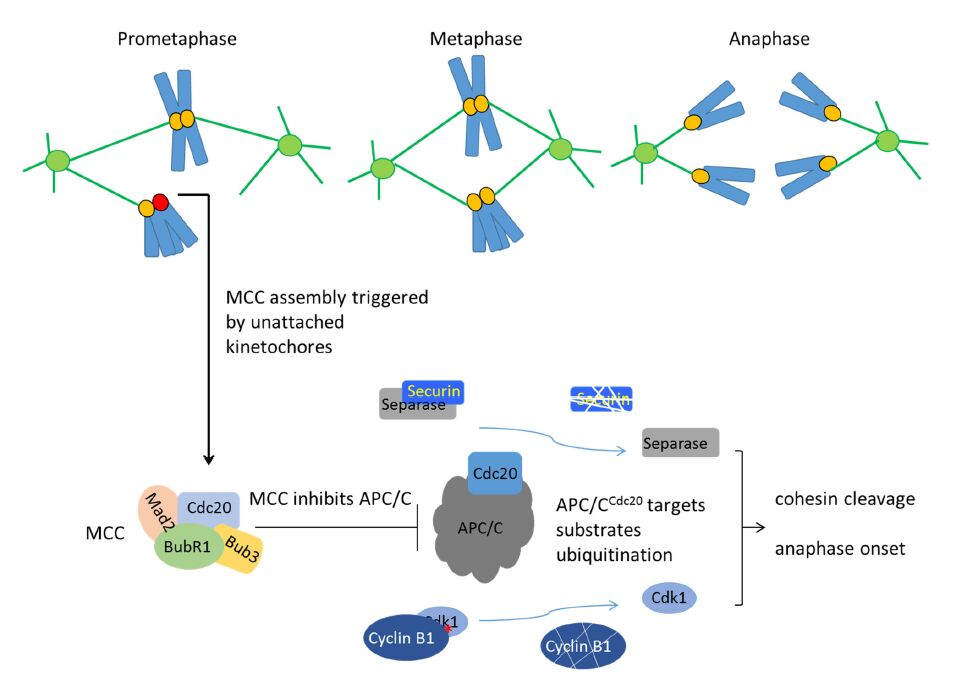
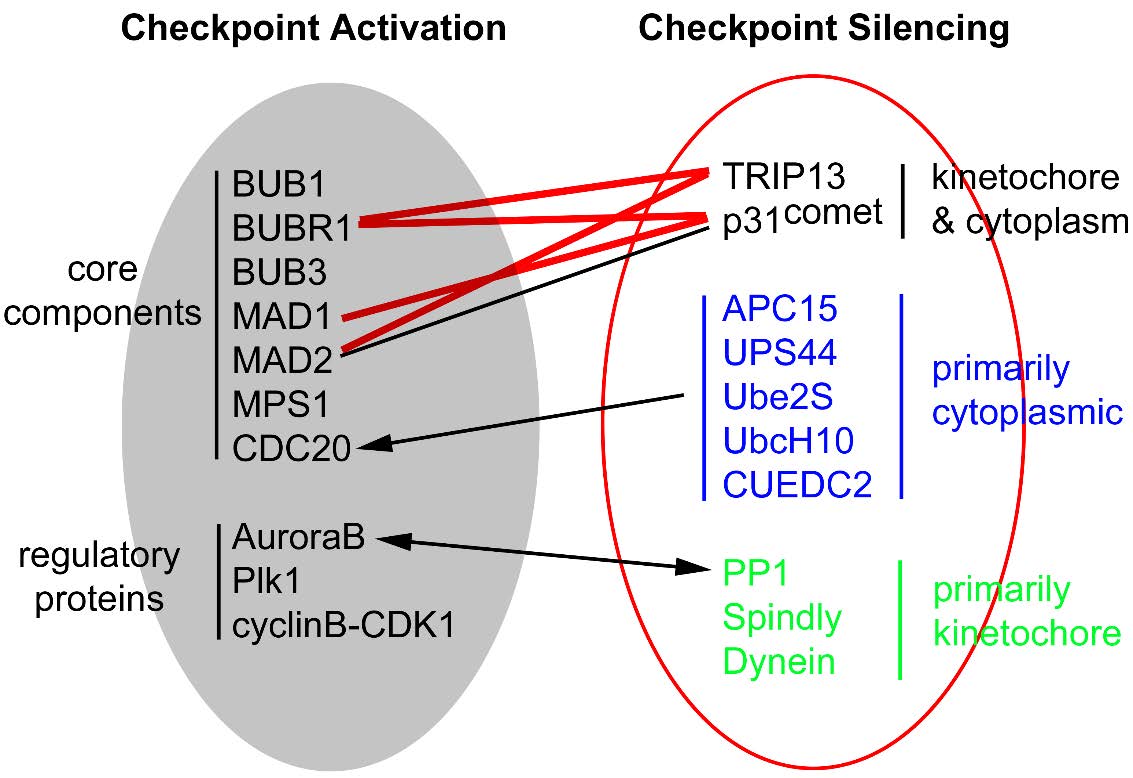
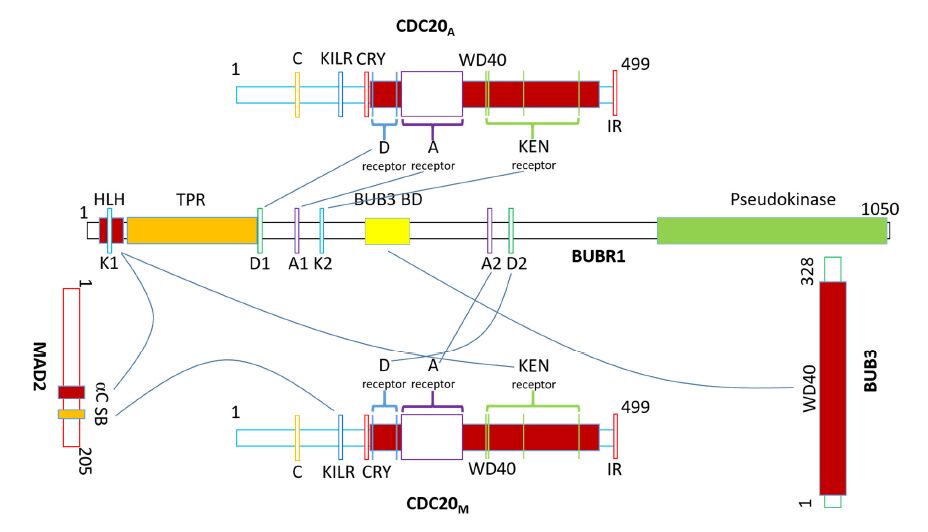
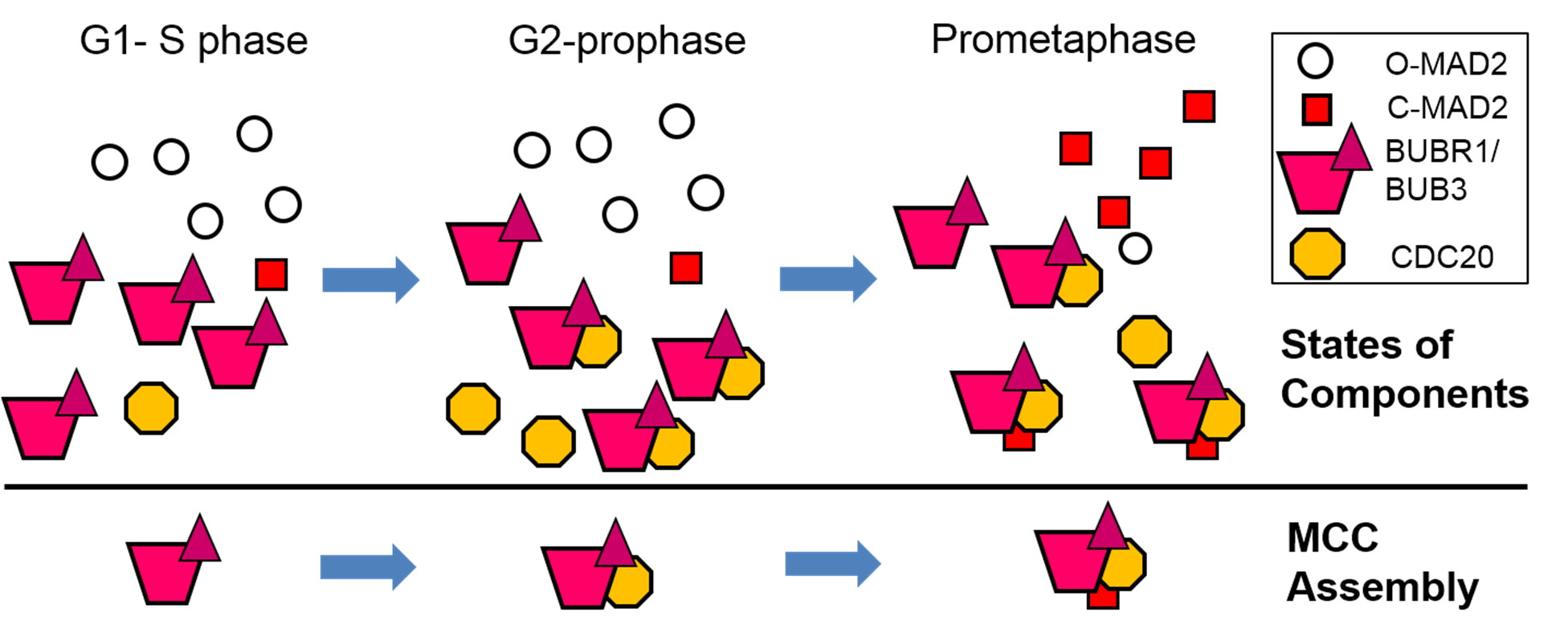
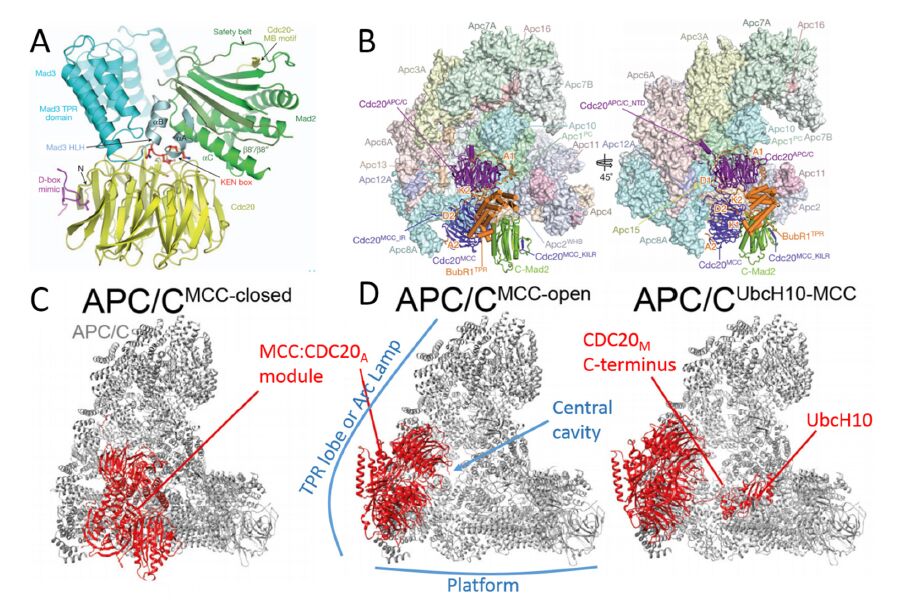
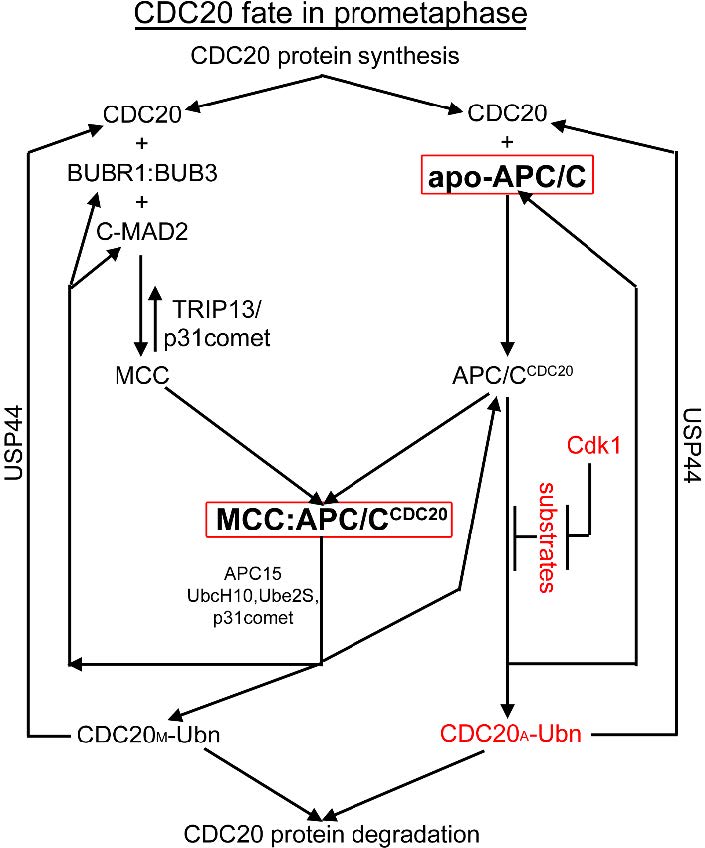
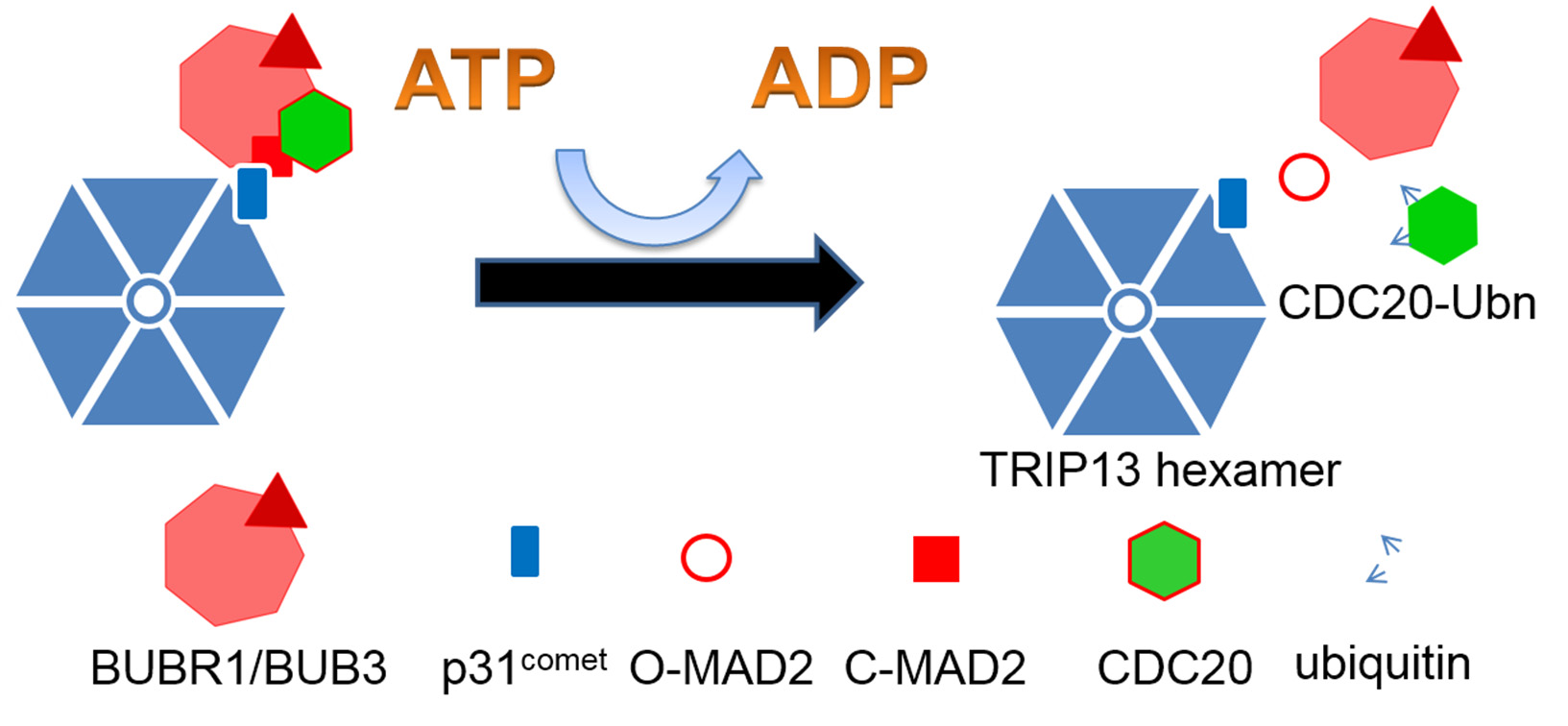
Figures(7) / Tables(2)

Song-Tao Liu, Hang Zhang. The mitotic checkpoint complex (MCC): looking back and forth after 15 years[J]. AIMS Molecular Science, 2016, 3(4): 597-634. doi: 10.3934/molsci.2016.4.597







 DownLoad:
DownLoad: