Citation: Hong-Yi Chang, Hung-Wen Tsai, Chiao-Fang Teng, Lily Hui-Ching Wang, Wenya Huang, Ih-Jen Su. Ground glass hepatocytes provide targets for therapy or prevention of hepatitis B virus-related hepatocellular carcinoma[J]. AIMS Medical Science, 2018, 5(2): 90-101. doi: 10.3934/medsci.2018.2.90

| [1] | Hadziyannis S, Gerber MA, Vissoulis C, et al. (1973) Cytoplasmic hepatitis B antigen in "ground-glass" hepatocytes of carriers. Arch Pathol 96: 327–330. |
| [2] |
Pópper H (1975) The ground glass hepatocyte as a diagnostic hint. Hum Pathol 6: 517–520. doi: 10.1016/S0046-8177(75)80069-4

|
| [3] | Shikata T (1973) Australia antigen in liver tissue-an immunofluorescent and immunoelectron microscopic study. Jpn J Exp Med 43: 231–245. |
| [4] | Shikata T, Uzawa T, Yoshiwara N, et al. (1974) Staining methods of Australia antigen in paraffin section-detection of cytoplasmic inclusion bodies. Jpn J Exp Med 44: 25–36. |
| [5] | Gudat F, Bianchi L, Sonnabend W, et al. (1975) Pattern of core and surface expression in liver tissue reflects state of specific immune response in hepatitis B. Lab Invest 32: 1–9. |
| [6] |
Hsu H, Lai M, Su I, et al. (1988) Correlation of hepatocyte HBsAg expression with virus replication and liver pathology. Hepatology 8: 749–754. doi: 10.1002/hep.1840080408
|
| [7] |
Wang HC, Wu HC, Chen CF, et al. (2003) Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am J Pathol 163: 2441–2449. doi: 10.1016/S0002-9440(10)63599-7
|
| [8] | Su IJ, Kuo TT, Liaw YF (1985) Hepatocyte hepatitis B surface antigen. Diagnostic evaluation of patients with clinically acute hepatitis B surface antigen-positive hepatitis. Arch Pathol Lab Med 109: 400–402. |
| [9] |
Hsu HC, Lin YH, Chang MH, et al. (1988) Pathology of chronic hepatitis B virus infection in children: with special reference to the intrahepatic expression of hepatitis B virus antigens. Hepatology 8: 378–382. doi: 10.1002/hep.1840080232
|
| [10] |
Fan YF, Lu CC, Chang YC, et al. (2000) Identification of a pre-S2 mutant in hepatocytes expressing a novel marginal pattern of surface antigen in advanced diseases of chronic hepatitis B virus infection. J Gastroenterol Hepatol 15: 519–528. doi: 10.1046/j.1440-1746.2000.02187.x
|
| [11] |
Su IJ, Lai MY, Hsu HC, et al. (1986) Diverse virological, histopathological and prognostic implications of seroconversion from hepatitis B e antigen to anti-HBe in chronic hepatitis B virus infection. J Hepatol 3: 182–189. doi: 10.1016/S0168-8278(86)80024-1
|
| [12] | Tai PC, Banik D, Lin GI, et al. (1997) Novel and frequent mutations of hepatitis B virus coincide with a major histocompatibility complex class I-restricted T-cell epitope of the surface antigen. J Virol 71: 4852–4856. |
| [13] | Hsu HC, Wu TT, Wu MZ, et al. (1988) Evolution of expression of hepatitis B surface and core antigens (HBsAg, HBcAg) in resected primary and recurrent hepatocellular carcinoma in HBsAg carriers in Taiwan. Correlation with local host immune response. Cancer 62: 915–921. |
| [14] | Mimms L (1995) Hepatitis B virus escape mutants: "Pushing the envelope" of chronic hepatitis B virus infection. Hepatology 21: 884–887. |
| [15] |
Hsieh YH, Su IJ, Wang HC, et al. (2007) Hepatitis B virus pre-S2 mutant surface antigen induces degradation of cyclin-dependent kinase inhibitor p27Kip1 through c-Jun activation domain-binding protein 1. Mol Cancer Res 5: 1063–1072. doi: 10.1158/1541-7786.MCR-07-0098
|
| [16] |
Liu S, Zhang H, Gu C, et al. (2009) Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: A meta-analysis. J Natl Cancer Inst 101: 1066–1082. doi: 10.1093/jnci/djp180
|
| [17] |
Santantonio T, Jung MC, Schneider R, et al. (1992) Hepatitis B virus genomes that cannot synthesize pre-S2 proteins occur frequently and as dominant virus populations in chronic carriers in Italy. Virology 188: 948–952. doi: 10.1016/0042-6822(92)90559-8
|
| [18] |
Fan YF, Lu CC, Chen WC, et al. (2001) Prevalence and significance of hepatitis B virus (HBV) pre-S mutants in serum and liver at different replicative stages of chronic HBV infection. Hepatology 33: 277–286. doi: 10.1053/jhep.2001.21163
|
| [19] |
Huy TT, Ushijima H, Win KM, et al. (2003) High prevalence of hepatitis B virus pre-S mutant in countries where it is endemic and its relationship with genotype and chronicity. J Clin Microbiol 41: 5449–5455. doi: 10.1128/JCM.41.12.5449-5455.2003
|
| [20] |
Fernholz D, Galle PR, Stemler M, et al. (1993) Infectious hepatitis B virus variant defective in pre-S2 protein expression in a chronic carrier. Virology 194: 137–148. doi: 10.1006/viro.1993.1243
|
| [21] | Le SJ, Chouteau P, Cannie I, et al. (1998) Role of the pre-S2 domain of the large envelope protein in hepatitis B virus assembly and infectivity. J Virol 72: 5573–5578. |
| [22] |
Su IJ, Wang HC, Wu HC, et al. (2008) Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J Gastroenterol Hepatol 23: 1169–1174. doi: 10.1111/j.1440-1746.2008.05348.x
|
| [23] |
Wang M, Kaufman RJ (2014) The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer 14: 581–597. doi: 10.1038/nrc3800
|
| [24] |
Hsieh YH, Su IH, Chang WW, et al. (2004) Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 25: 2023–2032. doi: 10.1093/carcin/bgh207
|
| [25] |
Hung JH, Su IJ, Lei HY, et al. (2004) Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J Biol Chem 279: 46384–46392. doi: 10.1074/jbc.M403568200
|
| [26] | Yen TT, Yang A, Chiu WT, et al. (2016) Hepatitis B virus PreS2-mutant large surface antigen activates store-operated calcium entry and promotes chromosome instability. Oncotarget 7: 23346–23360. |
| [27] |
Smyth JT, Hwang SY, Tomita T, et al. (2010) Activation and regulation of store-operated calcium entry. J Cell Mol Med 14: 2337–2349. doi: 10.1111/j.1582-4934.2010.01168.x
|
| [28] |
Wang LH, Huang W, Lai MD, et al. (2012) Aberrant cyclin A expression and centrosome overduplication induced by hepatitis B virus pre-S2 mutants and its implication in hepatocarcinogenesis. Carcinogenesis 33: 466–472. doi: 10.1093/carcin/bgr296
|
| [29] |
Wang HC, Chang WT, Chang WW, et al. (2005) Hepatitis B virus pre-S2 mutant upregulates cyclin A expression and induces nodular proliferation of hepatocytes. Hepatology 41: 761–770. doi: 10.1002/hep.20615
|
| [30] |
Sunami Y, Ringelhan M, Kokai E, et al. (2016) Canonical NF-kappaB signaling in hepatocytes acts as a tumor-suppressor in hepatitis B virus surface antigen-driven hepatocellular carcinoma by controlling the unfolded protein response. Hepatology 63: 1592–1607. doi: 10.1002/hep.28435
|
| [31] |
Hsieh YH, Chang YY, Su IJ, et al. (2015) Hepatitis B virus pre-S2 mutant large surface protein inhibits DNA double-strand break repair and leads to genome instability in hepatocarcinogenesis. J Pathol 236: 337–347. doi: 10.1002/path.4531
|
| [32] |
Hsieh YH, Hsu JL, Su IJ, et al. (2011) Genomic instability caused by hepatitis B virus: Into the hepatoma inferno. Front Biosci 16: 2586–2597. doi: 10.2741/3874
|
| [33] | Akbar SK, Onji M (1998) Hepatitis B virus (HBV)-transgenic mice as an investigative tool to study immunopathology during HBV infection. Int J Exp Pathol 79: 279–291. |
| [34] | Huang SN, Chisari FV (1995) Strong, sustained hepatocellular proliferation precedes hepatocarcinogenesis in hepatitis B surface antigen transgenic mice. Hepatology 21: 620–626. |
| [35] |
Hagen TM, Huang S, Curnutte J, et al. (1994) Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc Natl Acad Sci U S A 91: 12808–12812. doi: 10.1073/pnas.91.26.12808
|
| [36] |
Wang HC, Huang W, Lai MD, et al. (2006) Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci 97: 683–688. doi: 10.1111/j.1349-7006.2006.00235.x
|
| [37] |
Zheng Y, Chen WL, Louie SG, et al. (2007) Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice. Hepatology 45: 16–21. doi: 10.1002/hep.21445
|
| [38] |
Teng YC, Neo JC, Wu JC, et al. (2017) Expression of a hepatitis B virus pre-S2 deletion mutant in the liver results in hepatomegaly and hepatocellular carcinoma in mice. J Pathol 241: 463–474. doi: 10.1002/path.4850
|
| [39] |
Shen FC, Su IJ, Wu HC, et al. (2009) A pre-S gene chip to detect pre-S deletions in hepatitis B virus large surface antigen as a predictive marker for hepatoma risk in chronic hepatitis B virus carriers. J Biomed Sci 16: 84. doi: 10.1186/1423-0127-16-84
|
| [40] | Tsai HW, Lin YJ, Wu HC, et al. (2016) Resistance of ground glass hepatocytes to oral antivirals in chronic hepatitis B patients and implication for the development of hepatocellular carcinoma. Oncotarget 7: 27724–27734. |
| [41] |
Chang TT, Liaw YF, Wu SS, et al. (2010) Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology 52: 886–893. doi: 10.1002/hep.23785
|
| [42] |
Singal AK, Salameh H, Kuo YF, et al. (2013) Meta-analysis: The impact of oral anti-viral agents on the incidence of hepatocellular carcinoma in chronic hepatitis B. Aliment Pharmacol Ther 38: 98–106. doi: 10.1111/apt.12344
|
| [43] |
Wu CY, Chen YJ, Ho HJ, et al. (2012) Association between nucleoside analogues and risk of hepatitis B virus-related hepatocellular carcinoma recurrence following liver resection. JAMA 308: 1906–1914. doi: 10.1001/2012.jama.11975
|
| [44] |
Tsai HW, Lin YJ, Lin PW, et al. (2011) A clustered ground-glass hepatocyte pattern represents a new prognostic marker for the recurrence of hepatocellular carcinoma after surgery. Cancer 117: 2951–2960. doi: 10.1002/cncr.25837
|
| [45] | Manesis EK, Hadziyannis ES, Angelopoulou OP, et al. (2007) Prediction of treatment-related HBsAg loss in HBeAG-negative chronic hepatitis B: A clue from serum HBsAg levels. Antiviral Ther 12: 73–82. |
| [46] |
Wong DK, Cheung AM, O'Rourke K, et al. (1993) Effect of alpha-interferon treatment in patients with hepatitis B e antigen-positive chronic hepatitis B. A meta-analysis. Ann Intern Med 119: 312–323. doi: 10.7326/0003-4819-119-4-199308150-00011
|
| [47] |
Teng CF, Wu HC, Hsieh WC, et al. (2015) Activation of ATP citrate lyase by mTOR signal induces disturbed lipid metabolism in hepatitis B virus pre-S2 mutant tumorigenesis. J Virol 89: 605–614. doi: 10.1128/JVI.02363-14
|
| [48] |
Loguercio C, Andreone P, Brisc C, et al. (2012) Silybin combined with phosphatidylcholine and vitamin E in patients with nonalcoholic fatty liver disease: A randomized controlled trial. Free Radical Biol Med 52: 1658–1665. doi: 10.1016/j.freeradbiomed.2012.02.008
|
| [49] |
Polyak SJ, Morishima C, Lohmann V, et al. (2010) Identification of hepatoprotective flavonolignans from silymarin. Proc Natl Acad Sci U S A 107: 5995–5999. doi: 10.1073/pnas.0914009107
|
| [50] |
Jang M, Cai L, Udeani GO, et al. (1997) Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275: 218–220. doi: 10.1126/science.275.5297.218
|
| [51] |
Gusman J, Malonne H, Atassi G (2001) A reappraisal of the potential chemopreventive and chemotherapeutic properties of resveratrol. Carcinogenesis 22: 1111–1117. doi: 10.1093/carcin/22.8.1111
|
| [52] |
Fulda S (2010) Resveratrol and derivatives for the prevention and treatment of cancer. Drug Discovery Today 15: 757–765. doi: 10.1016/j.drudis.2010.07.005
|
| [53] |
Guarente L (2006) Sirtuins as potential targets for metabolic syndrome. Nature 444: 868–874. doi: 10.1038/nature05486
|
| [54] |
Lekli I, Ray D, Das DK (2010) Longevity nutrients resveratrol, wines and grapes. Genes Nutr 5: 55–60. doi: 10.1007/s12263-009-0145-2
|
| [55] |
Teng CF, Hsieh WC, Wu HC, et al. (2015) Hepatitis B Virus Pre-S2 Mutant Induces Aerobic Glycolysis through Mammalian Target of Rapamycin Signal Cascade. PLoS One 10: e0122373. doi: 10.1371/journal.pone.0122373
|
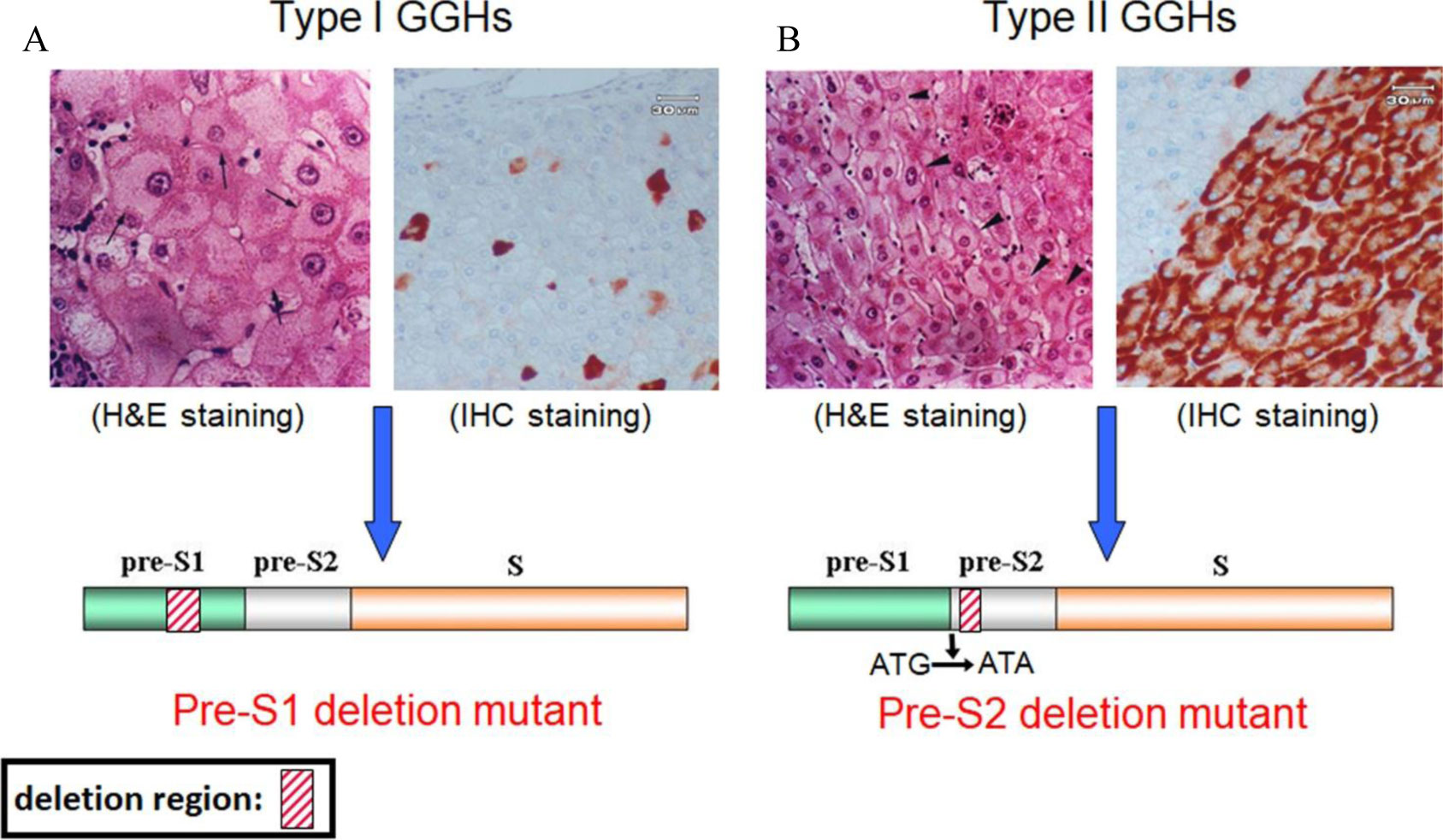
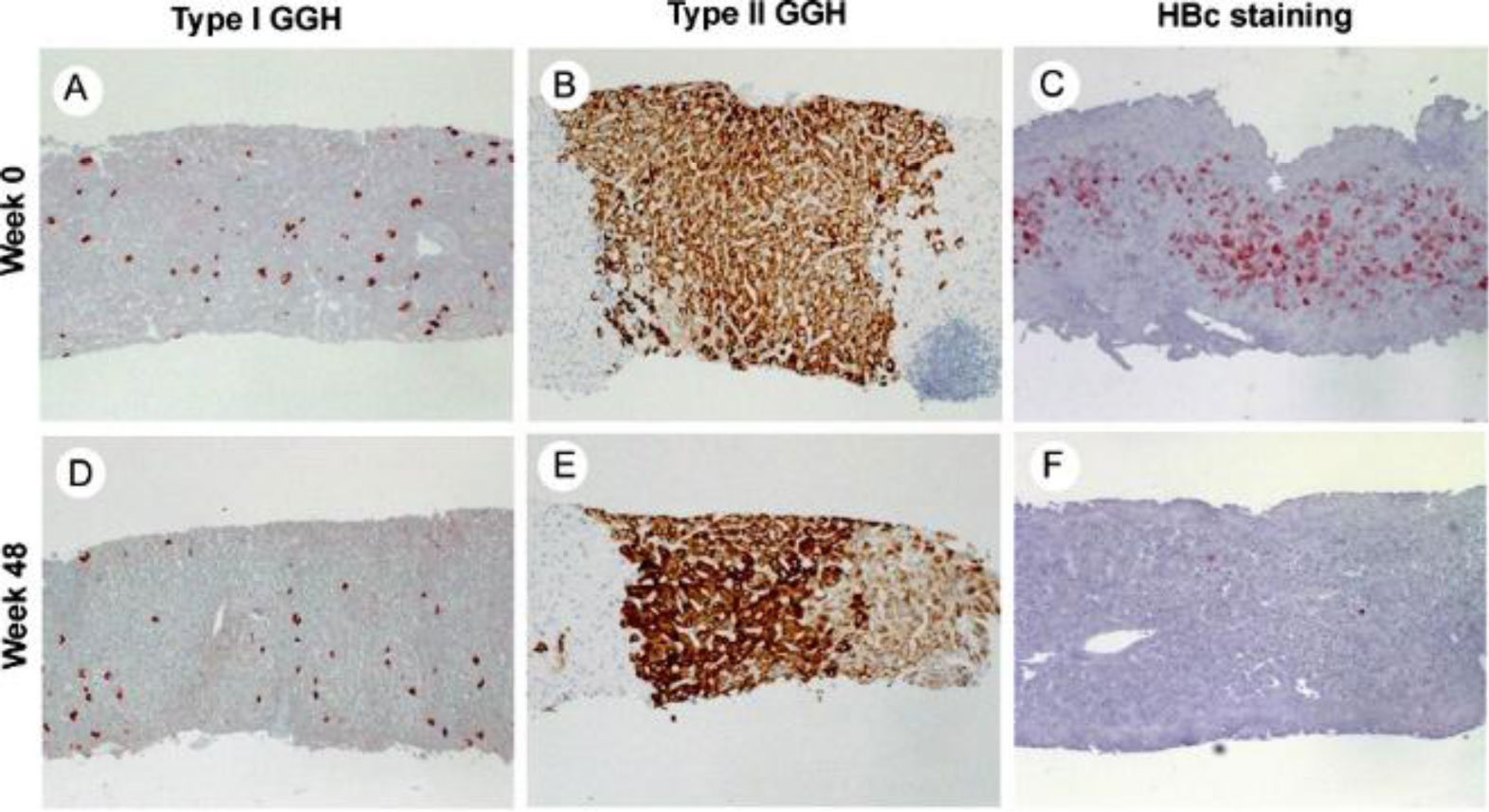
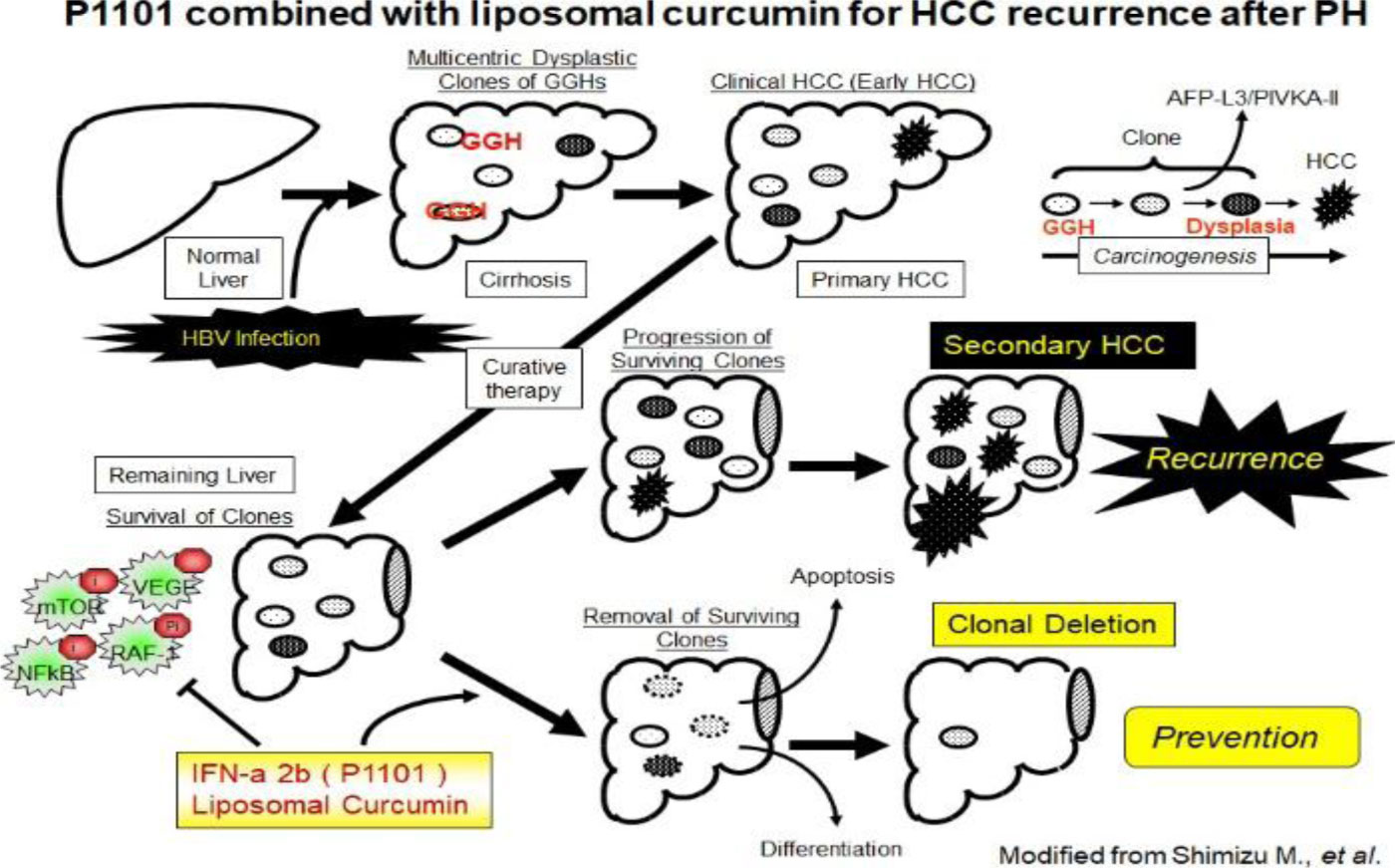
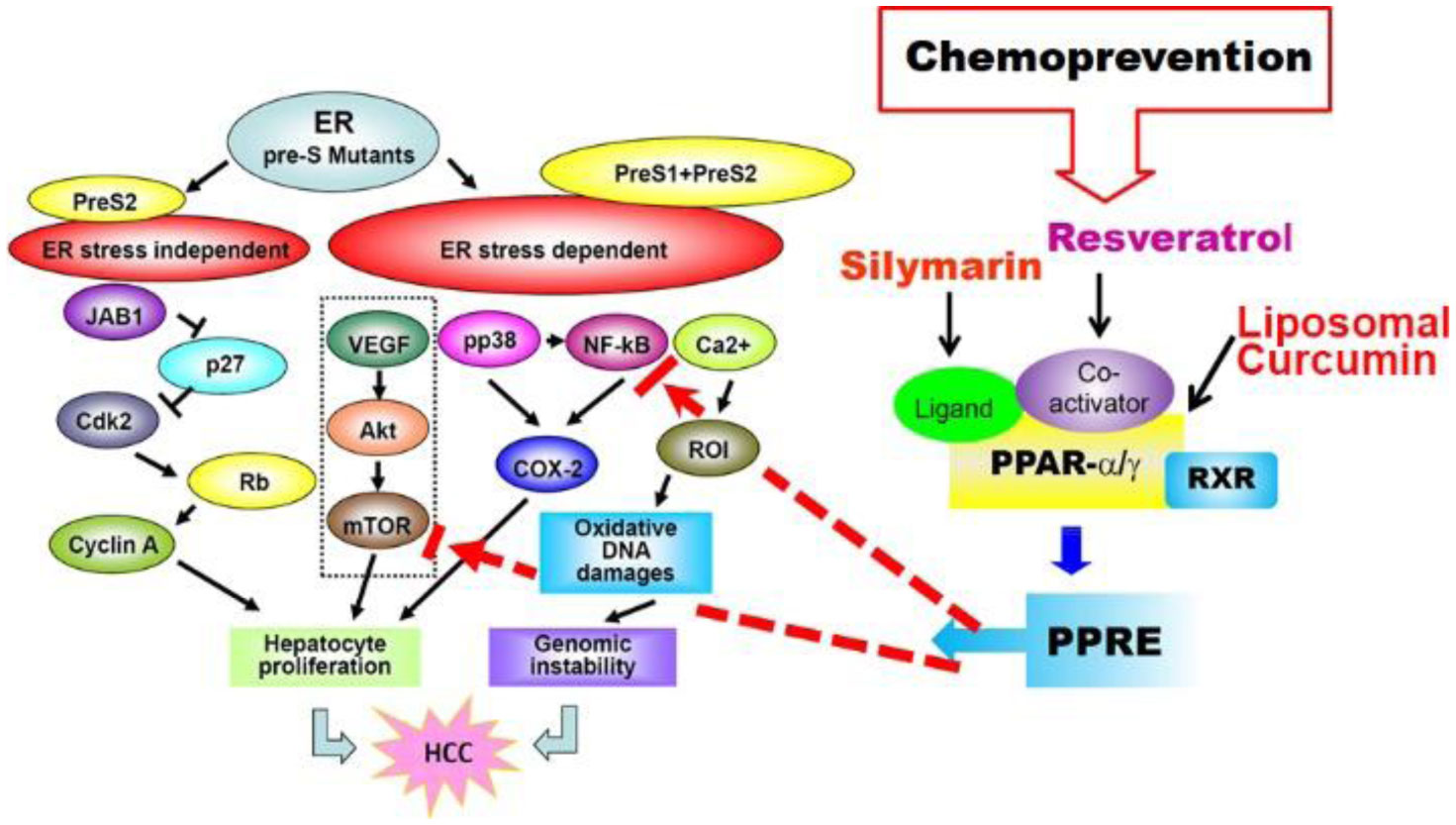
Figures(4)

Hong-Yi Chang, Hung-Wen Tsai, Chiao-Fang Teng, Lily Hui-Ching Wang, Wenya Huang, Ih-Jen Su. Ground glass hepatocytes provide targets for therapy or prevention of hepatitis B virus-related hepatocellular carcinoma[J]. AIMS Medical Science, 2018, 5(2): 90-101. doi: 10.3934/medsci.2018.2.90







 DownLoad:
DownLoad: