Citation: Tyson Loudon, Stephen Pankavich. Mathematical analysis and dynamic active subspaces for a long term model of HIV[J]. Mathematical Biosciences and Engineering, 2017, 14(3): 709-733. doi: 10.3934/mbe.2017040

| [1] | [ D. Callaway,A. Perelson, HIV-1 infection and low steady state viral loads, Bull.Math.Biol., 64 (2002): 29-64. |
| [2] | [ P. Constantine, Active Subspaces: Emerging Ideas for Dimension Reduction in Parameter Studies SIAM, 2015. |
| [3] | [ P. Constantine and D. Gleich, Computing active subspaces with monte carlo, arXiv: 1408.0545 |
| [4] | [ P. Constantine,B. Zaharatos,M. Campanelli, Discovering an active subspace in a single-diode solar cell model, Statistical Analysis and Data Mining: The ASA Data Science Journal, 8 (2015): 264-273. |
| [5] | [ A. S. Fauci,G. Pantaleo,S. Stanley, Immunopathogenic mechanisms of HIV infection, Annals of Internal Medicine, 124 (1996): 654-663. |
| [6] | [ T. C. Greenough,D. B. Brettler,F. Kirchhoff, Long-term non-progressive infection with Human Immunodeficiency Virus in a Hemophilia cohort, J Infect Dis, 180 (1999): 1790-1802. |
| [7] | [ A. B. Gumel,P. N. Shivakumar,B. M. Sahai, A mathematical model for the dynamics of HIV-1 during the typical course of infection, Nonlinear Analysis, 47 (2001): 1773-1783. |
| [8] | [ M. Hadjiandreou,R. Conejeros,V. S. Vassiliadis, Towards a long-term model construction for the dynamic simulation of HIV infection, Mathematical Biosciences and Engineering, 4 (2007): 489-504. |
| [9] | [ E. Hernandez-Vargas,R. Middleton, Modeling the three stages in HIV infection, J TheorBiol., 320 (2013): 33-40. |
| [10] | [ T. Igarashi,C. R. Brown,Y. Endo, Macrophages are the principal reservoir and sustain high virus loads in Rhesus Macaques following the depletion of CD4+ T-cells by a highly pathogenic SIV: Implications for HIV-1 infections of man, Proc Natl Acad Sci., 98 (2001): 658-663. |
| [11] | [ E. Jones and P. Roemer (sponsors: S. Pankavich and M. Raghupathi), Analysis and simulation of the three-component model of HIV dynamics, SIAM Undergraduate Research Online, 7 (2014), 89–106 |
| [12] | [ D. Kirschner, Using mathematics to understand HIV immunodynamics, Am. Math. Soc., 43 (1996): 191-202. |
| [13] | [ D. E. Kirschner and A. S. Perelson, A model for the immune response to HIV: AZT treatment studies, Mathematical Population Dynamics: Analysis of Heterogeneity, Volume One: Theory of Epidemics Eds. O. Arino, D. Axelrod, M. Kimmel, and M. Langlais, Wuerz Publishing Ltd., Winnipeg, Canada, (1993), 295–310. |
| [14] | [ D. Kirschner,G. F. Webb, Immunotherapy of HIV-1 infection, J Biological Systems, 6 (1998): 71-83. |
| [15] | [ D. Kirschner,G. F. Webb,M. Cloyd, A model of HIV-1 disease progression based on virus-induced lymph node homing-induced apoptosis of CD4+ lymphocytes, J Acquir Immune Dec Syndr, 24 (2000): 352-362. |
| [16] | [ J. M. Murray,G. Kaufmann,A. D. Kelleher, A model of primary HIV-1 infection, Math Biosci, 154 (1998): 57-85. |
| [17] | [ M. Nowak and R. May, Virus Dynamics: Mathematical Principles of Immunology and Virology Oxford University Press, NewYork, 2000. |
| [18] | [ S. Pankavich, The effects of latent infection on the dynamics of HIV, Differential Equations and Dynamical Systems, 24 (2016): 281-303. |
| [19] | [ S. Pankavich,D. Shutt, An in-host model of HIV incorporating latent infection and viral mutation, Dynamical Systems, Differential Equations, and Applications, AIMS Proceedings, null (2015): 913-922. |
| [20] | [ S. Pankavich, N. Neri and D. Shutt, Bistable dynamics and Hopf bifurcation in a refined model of the acute stage of HIV infection, submitted, (2015). |
| [21] | [ S. Pankavich,C. Parkinson, Mathematical analysis of an in-host model of viral dynamics with spatial heterogeneity, Discrete and Continuous Dynamical Systems B, 21 (2016): 1237-1257. |
| [22] | [ E. Pennisi and J. Cohen, Eradicating HIV from a patient: Not just a dream?, Science, 272 (1996), 1884. |
| [23] | [ A. S. Perelson, Modeling the Interaction of the Immune System with HIV, Lecture Notes in Biomath. Berlin: Springer, 1989. |
| [24] | [ A. Perelson,P. Nelson, Mathematical analysis of HIV-1 dynamics in vivo, SIAM Rev., 41 (1999): 3-44. |
| [25] | [ T. M. Russi, Uncertainty Quantification with Experimental data and Complex System Models, Ph. D. thesis, UC Berkeley, 2010. |
| [26] | [ W. Y. Tan,H. Wu, Stochastic modeing of the dynamics of CD4+ T-cell infection by HIV and some monte carlo studies, Math Biosci, 147 (1997): 173-205. |
| [27] | [ E. Vergu,A. Mallet,J. Golmard, A modeling approach to the impact of HIV mutations on the immune system, Comput Biol Med., 35 (2005): 1-24. |
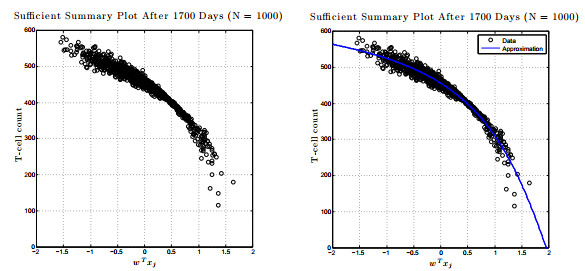
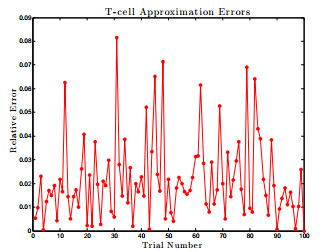
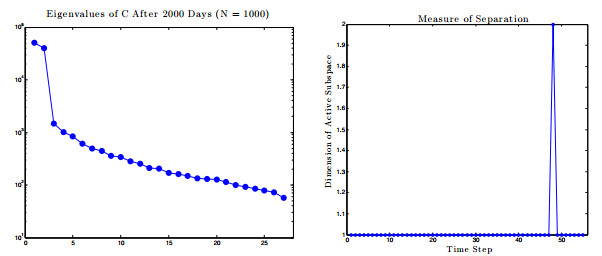

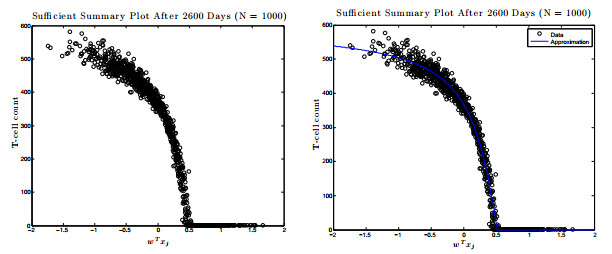


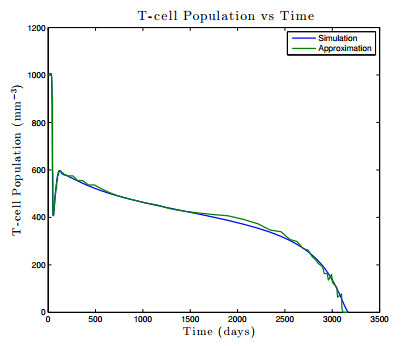
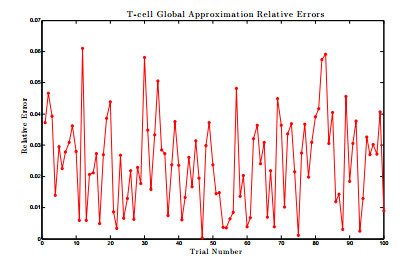
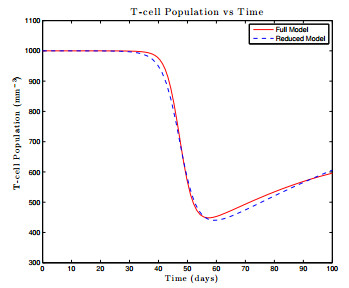
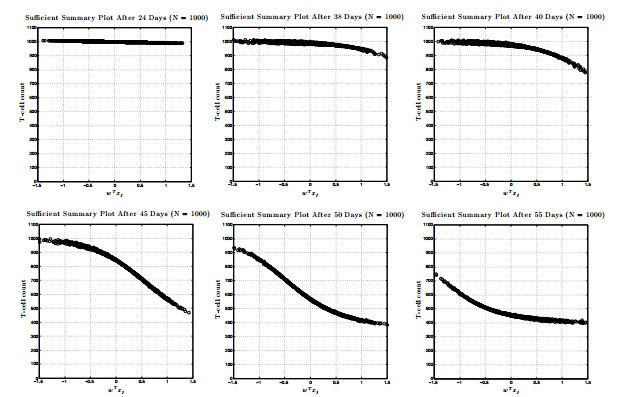

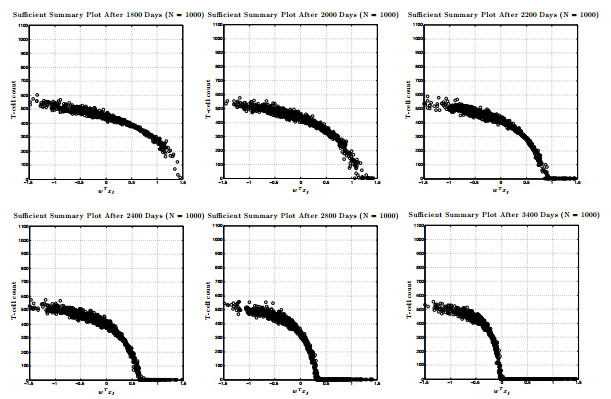
Figures(17) / Tables(1)

Tyson Loudon, Stephen Pankavich. Mathematical analysis and dynamic active subspaces for a long term model of HIV[J]. Mathematical Biosciences and Engineering, 2017, 14(3): 709-733. doi: 10.3934/mbe.2017040







 DownLoad:
DownLoad: