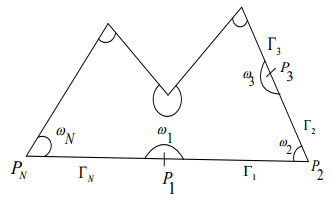
This study typically emphasizes analyzing the geometrical singularities of weak solutions of the mixed boundary value problem for the stationary Stokes and Navier-Stokes system in two-dimensional non-smooth domains with corner points and points at which the type of boundary conditions change. The existence of these points on the boundary generally generates local singularities in the solution. We will see the impact of the geometrical singularities of the boundary or the mixed boundary conditions on the qualitative properties of the solution including its regularity. The solvability of the underlying boundary value problem is analyzed in weighted Sobolev spaces and the regularity theorems are formulated in the context of these spaces. To compute the singular terms for various boundary conditions, the generalized form of the boundary eigenvalue problem for the stationary Stokes system is derived. The emerging eigenvalues and eigenfunctions produce singular terms, which permits us to evaluate the optimal regularity of the corresponding weak solution of the Stokes system. Additionally, the obtained results for the Stokes system are further extended for the non-linear Navier-Stokes system.
Citation: Yasir Nadeem Anjam. The qualitative analysis of solution of the Stokes and Navier-Stokes system in non-smooth domains with weighted Sobolev spaces[J]. AIMS Mathematics, 2021, 6(6): 5647-5674. doi: 10.3934/math.2021334
This study typically emphasizes analyzing the geometrical singularities of weak solutions of the mixed boundary value problem for the stationary Stokes and Navier-Stokes system in two-dimensional non-smooth domains with corner points and points at which the type of boundary conditions change. The existence of these points on the boundary generally generates local singularities in the solution. We will see the impact of the geometrical singularities of the boundary or the mixed boundary conditions on the qualitative properties of the solution including its regularity. The solvability of the underlying boundary value problem is analyzed in weighted Sobolev spaces and the regularity theorems are formulated in the context of these spaces. To compute the singular terms for various boundary conditions, the generalized form of the boundary eigenvalue problem for the stationary Stokes system is derived. The emerging eigenvalues and eigenfunctions produce singular terms, which permits us to evaluate the optimal regularity of the corresponding weak solution of the Stokes system. Additionally, the obtained results for the Stokes system are further extended for the non-linear Navier-Stokes system.

| [1] | R. A. Adams, J. J. Fournier. Sobolev spaces, Vol. 140, Academic Press, 2003. |
| [2] |
S. Agmon, A. Douglis, L. Nirenberg, Estimates near the boundary for solutions of elliptic partial differential equations satisfying general boundary conditions I, Commun. Pure Appl. Math., 12 (1959), 623–727. doi: 10.1002/cpa.3160120405

|
| [3] |
S. Agmon, A. Douglis, L. Nirenberg, Estimates near the boundary for solutions of elliptic partial differential equations satisfying general boundary conditions II, Commun. Pure Appl. Math., 17 (1964), 35–92. doi: 10.1002/cpa.3160170104
|
| [4] |
Y. N. Anjam, Singularities and regularity of stationary Stokes and Navier-Stokes equations on polygonal domains and their treatments, AIMS Mathematics, 5 (2020), 440–466. doi: 10.3934/math.2020030
|
| [5] |
I. Babu$\check{s}$ka, B. Q. Guo, J. E. Osborn, Regularity and numerical solution of eigenvalue problems with piecewise analytic data, SIAM J. Num. Comp., 26 (1989), 1534–1560. doi: 10.1137/0726090
|
| [6] |
M. Bene$\check{{s}}$, P. Ku$\check{{c}}$era, Solutions of the Navier-Stokes equations with various types of boundary conditions, Arch. Math., 98 (2012), 487–497. doi: 10.1007/s00013-012-0387-x
|
| [7] |
H. J. Choi, J. R. Kweon, The stationary Navier-Stokes system with no-slip boundary condition on polygons: corner singularity and regularity, Commun. Part. Diff. Eq., 38 (2013), 1235–1255. doi: 10.1080/03605302.2012.752386
|
| [8] | N. Chorfi, Geometric singularities of the Stokes problem, Abstr. Appl. Anal., 2014 (2014), 1–8. |
| [9] | P. G. Ciarlet, Mathematical elasticity (studies in mathematics and its applications), Elsevier, 1988. |
| [10] |
M. Dauge, Stationary Stokes and Navier-Stokes systems on two or three-dimensional domains with corners. part I. linearized equations, SIAM J. Math. Anal., 20 (1989), 74–97. doi: 10.1137/0520006
|
| [11] | M. Dauge, Elliptic boundary value problems on corner domains: smoothness and asymptotics of solutions, Vol. 1341, 2006. |
| [12] | M. Durand, Singularities in elliptic problems. In: Singularities and constructive methods for their treatment, Springer, Berlin, Heidelberg, 1985. |
| [13] |
J. Fabricius, Stokes flow with kinematic and dynamic boundary conditions, Quart. Appl. Math., 77 (2019), 525–544. doi: 10.1090/qam/1534
|
| [14] | S. Fucik, O. John, A. Kufner, Function spaces, Springer, Netherlands, 1977. |
| [15] | V. Girault, P. A. Raviart, Finite element methods for Navier-Stokes equations: theory and algorithms, Vol. 5, Springer Science and Business Media, 2012. |
| [16] | P. Grisvard, Behavior of the solutions of an elliptic boundary value problem in a polygonal or polyhedral domain, Numerical solution of partial differential equations, III, (1976), 207–274. |
| [17] | P. Grisvard, Elliptic problems in nonsmooth domains, Vol. 2, 2–2, Pitman Advanced Pub. Program, Boston, 1985. |
| [18] |
B. Q. Guo, I. Babu$\check{s}$ka, On the regularity of elasticity problems with piecewise analytic data, Adv. Appl. Math., 14 (1993), 307–347. doi: 10.1006/aama.1993.1016
|
| [19] |
B. Q. Guo, C. Schwab, Analytic regularity of Stokes flow on polygonal domains in countably weighted Sobolev spaces, J. Comput. Appl. Math., 190 (2006), 487–519. doi: 10.1016/j.cam.2005.02.018
|
| [20] |
Y. Hou, S. Pei, On the weak solutions to steady Navier-Stokes equations with mixed boundary conditions, Math. Zeit., 291 (2019), 47–54. doi: 10.1007/s00209-018-2072-7
|
| [21] |
S. Itoh, N. Tanaka, A. Tani, On some boundary value problem for the stokes equations in an infinite sector, Anal. Appl., 4 (2006), 357–375. doi: 10.1142/S0219530506000826
|
| [22] | V. V. Katrakhov, S. V. Kiselevskaya, A singular elliptic boundary value problem in domains with corner points. I. Function spaces, Diff. Eq., 42 (2006), 395–403. |
| [23] |
R. B. Kellogg, J. E. Osborn, A regularity result for the Stokes problem in a convex polygon, J. Func. Anal., 21 (1976), 397–431. doi: 10.1016/0022-1236(76)90035-5
|
| [24] | V. A. Kondrat$\acute{i}$ev, Boundary value problems for elliptic equations in domains with conical or angular points, Trudy Mos. Matem. Obsh., 16 (1967), 209–292. |
| [25] | V. A. Kondrat$\acute{i}$ev, O. A. Oleinik, Boundary-value problems for partial differential equations in non-smooth domains, Russ. Math. Surveys, 38 (1983), 1–86. |
| [26] | V. A. Kozlov, V. G. Maz'ya, J. Rossmann, Elliptic boundary value problems in domains with point singularities, American Mathematical Society, 1997. |
| [27] | V. A. Kozlov, V. G. Maz'ya, J. Rossmann, Spectral problems associated with corner singularities of solutions to elliptic equations, Vol. 85, American Mathematical Society, 2001. |
| [28] |
S. Kr$\breve{a}$cmar, J. Neustupa, A weak solvability of a steady variational inequality of the Navier-Stokes type with mixed boundary conditions, Nonlinear Anal-Theor, 47 (2001), 4169–4180. doi: 10.1016/S0362-546X(01)00534-X
|
| [29] | S. G. Krejn, V. P. Trofimov, Holomorphic operator-valued functions of several complex variables, Funct. Anal. i Prilo$\breve{z}$en., 3 (1969), 85–86. |
| [30] |
P. Ku$\check{c}$era, Basic properties of solution of the non-steady Navier-Stokes equations with mixed boundary conditions in a bounded domain, Annali dell' Univ. di Ferrara. Sezione VII. Scie. Matem., 55 (2009), 289–308. doi: 10.1007/s11565-009-0082-4
|
| [31] |
J. R. Kweon, Regularity of solutions for the Navier-Stokes system of incompressible flows on a polygon, J. Diff. Eq., 235 (2007), 166–198. doi: 10.1016/j.jde.2006.12.008
|
| [32] |
J. R. Kweon, Edge singular behavior for the heat equation on polyhedral cylinders in $R^{3}$, Potential Anal., 38 (2013), 589–610. doi: 10.1007/s11118-012-9288-7
|
| [33] |
J. R. Kweon, The compressible Stokes flows with no-slip boundary condition on non-convex polygons, J. Math. Fluid Mech., 19 (2017), 47–57. doi: 10.1007/s00021-016-0264-7
|
| [34] |
O. S. Kwon, J. R. Kweon, Compressible Navier-Stokes equations in a polyhedral cylinder with inflow boundary condition, J. Math. Fluid Mech., 20 (2018), 581–601. doi: 10.1007/s00021-017-0336-3
|
| [35] |
I. Lasiecka, K. Szulc, A. $\dot{Z}$ochowski, Boundary control of small solutions to fluid-structure interactions arising in coupling of elasticity with Navier-Stokes equation under mixed boundary conditions, Nonlinear Anal-Real, 44 (2018), 54–85. doi: 10.1016/j.nonrwa.2018.04.004
|
| [36] |
V. Maza, J. Rossmann, Mixed boundary value problems for the stationary Navier-Stokes system in polyhedral domains, Arch. Rati. Mech. Anal., 194 (2009), 669–712. doi: 10.1007/s00205-008-0171-z
|
| [37] |
S. A. Nazarov, A. Novotny, K. Pileckas, On steady compressible Navier-Stokes equations in plane domains with corners, Math. Ann., 304 (1996), 121–150. doi: 10.1007/BF01446288
|
| [38] | M. Orlt, A. M. S$\ddot{a}$ndig, Regularity of viscous Navier-Stokes flows in nonsmooth domains. Boundary value problems and integral equations in nonsmooth domains, Lecture Notes in Pure and Applied Mathematics, 167 (1995), 185–201. |
| [39] | A. M. S$\ddot{a}$ndig, Some applications of weighted Sobolev spaces, Vieweg+Teubner Verlag, 1987. |
| [40] | A. E. Taylor, Introduction to functional analysis, John Wiley and Sons, London, 1958. |
| [41] | R. Temam, Navier-Stokes equations: Theory and numerical analysis, Elsevier North-Holland, 1979. |



Figures(4)

Yasir Nadeem Anjam. The qualitative analysis of solution of the Stokes and Navier-Stokes system in non-smooth domains with weighted Sobolev spaces[J]. AIMS Mathematics, 2021, 6(6): 5647-5674. doi: 10.3934/math.2021334







 DownLoad:
DownLoad: