One of the most significant and growing negative pressures on the environment is the amount of generated waste. Surprisingly, the literature on this topic is mainly focused on monitoring the waste weight instead of volume, which is crucial for waste management in urban space. This paper is dedicated to this issue in two ways. First of all the study shows the difference in waste generation rates monitored in Poznań - a major city in Poland, during one year of field research focused on waste volume and its relationship to administrative data about waste weight. Secondly, it describes the volume-weight relationship based on the collection point sensors and a dynamic weighing system installed on a garbage truck and proposes a more accurate weight to volume conversion method.
Citation: Patrycja Przewoźna, Piotr Jankowski, Alfred Stach. Solid waste management in urban space: the volume-weight relationship[J]. AIMS Environmental Science, 2020, 7(6): 575-588. doi: 10.3934/environsci.2020036
One of the most significant and growing negative pressures on the environment is the amount of generated waste. Surprisingly, the literature on this topic is mainly focused on monitoring the waste weight instead of volume, which is crucial for waste management in urban space. This paper is dedicated to this issue in two ways. First of all the study shows the difference in waste generation rates monitored in Poznań - a major city in Poland, during one year of field research focused on waste volume and its relationship to administrative data about waste weight. Secondly, it describes the volume-weight relationship based on the collection point sensors and a dynamic weighing system installed on a garbage truck and proposes a more accurate weight to volume conversion method.

| [1] |
Beigl P, Lebersorger S, Salhofer S (2008) Modelling municipal solid waste generation: A review. Waste Manag 28: 200–214. doi: 10.1016/j.wasman.2006.12.011

|
| [2] |
den Boer E, Jędrczak A, Kowalski Z, et al. (2010) A review of municipal solid waste composition and quantities in Poland. Waste Manag 30: 367–377. doi: 10.1016/j.wasman.2009.11.020
|
| [3] |
Kolekar KA, Hazra T, Chakrabarty SN (2016) A Review on Prediction of Municipal Solid Waste Generation Models. Procedia Environ Sci 35:238–244. doi: 10.1016/j.proenv.2016.07.087
|
| [4] |
Emery AD, Griffiths AJ, Williams KP (2003) An in depth study of the effects of socio-economic conditions on household waste recycling practices. Waste Manag Res 21: 180–190. doi: 10.1177/0734242X0302100302
|
| [5] | Mamełka D (2008) Sprawozdanie z poboru prób oraz z przeprowadzonych badań morfologicznych i własności technicznych odpadów komunalnych z terenu Miasta Poznania w okresie październik - grudzień 2007 oraz kwiecień - maj 2008r. (Report on sampling and on morphological tests and technical properties of municipal waste from the city of Poznań in October - December 2007 and April - May 2008), Warsaw: Miej. Lab. Chem. przy Urzȩdzie m.st. Warszawy. |
| [6] | Dennison GJ, Dodd VA, Whelan B (1996) A socio-economic based survey of household waste characteristics in the city of Dublin, Ireland. I. Waste composition. Resour Conserv Recycl 17: 227–244. |
| [7] | Dennison GJ, Dodd VA, Whelan B (1996) A socio-economic based survey of household waste characteristics in the city of Dublin, Ireland. Ⅱ. Waste Quantities. Resour Conserv Recycl 17: 245–257. |
| [8] | LXiao L, Lin T, Chen S, et al. (2015) Characterizing Urban Household Waste Generation and Metabolism Considering Community Stratification in a Rapid Urbanizing Area of China. PLoS One 10: 1–16. |
| [9] |
Elia V, Gnoni MG, Tornese F (2015) Designing Pay-As-You-Throw schemes in municipal waste management services: A holistic approach. Waste Manag 44: 188–195. doi: 10.1016/j.wasman.2015.07.040
|
| [10] | Center for Sustainable Systems University of Michigan (2018) Municipal Solid Waste Factsheet Pub. No. CSS04-15. Available from: http://css.umich.edu/factsheets/municipal-solid-waste-factsheet. |
| [11] | Eurostat - Statistical Office of the European Communities (2015) Waste statistics. Available from: http://ec.europa.eu/eurostat/statistics-explained/index.php/Waste_statistics. |
| [12] | Jȩdrczak A, Szpadt R (2006) Określenie metodyki badań składu sitowego, morfologicznego i chemicznego odpadów komunalnych (Specific methods for testing the sieve, morphological and chemical composition of municipal waste). Narodowy Fundusz Ochrony Środowiska i Gospodarki Wodnej na zamówienie Ministra Środowiska, Kamieniec Wr. - Zielona Góra. |
| [13] | Bilitewski B, Wagner J, Reichenbach J (2018) Best Practice Municipal Waste Management. Umweltbundesamt, Available from: https://www.umweltbundesamt.de/en/publikationen/best-practice-municipal-waste-management. |
| [14] |
de Souza Melaré AV, González SM, Faceli K, et al. (2017) Technologies and decision support systems to aid solid-waste management: a systematic review. Waste Manag 59: 567–584. doi: 10.1016/j.wasman.2016.10.045
|
| [15] |
Al Mamun MA, Hannan MA, Hussain A (2014) A Novel Prototype and Simulation Model for Real Time Solid Waste Bin Monitoring System. J Kejuruter 26: 15–19. doi: 10.17576/jkukm-2014-26-02
|
| [16] |
Gutierrez JM, Jensen M, Henius M, et al. (2015) Smart Waste Collection System Based on Location Intelligence. Procedia Comput Sci 61: 120–127. doi: 10.1016/j.procs.2015.09.170
|
| [17] | Hassan SA, Jameel NGM, Şekeroğlu B (2016) Smart Solid Waste Monitoring and Collection System. Int J Adv Res Comput Sci Softw Eng Sci Softw Eng 6: 7–12. |
| [18] | Ramani H, Mehra L (2016) Technologies and Their Usage in Solid Waste Monitoring and Management Systems. National Conference on Road Map for Smart Cities of Rajasthan (NC-RMSCR) (Conference proceedings): 350–353. |
| [19] | Bani MS, Rashid ZA, Hamid KHK, et al. (2009) The development of decision support system for waste management; a review. World Acad Sci Eng Technol 37: 161–168. |
| [20] | Stępień M, Białecka B, Stalmachova B (2018) IT Systems Supporting Waste Management in Municipalities – Research Results. Multidiscip Asp Prod Eng 1: 777–783. |
| [21] | Wysocka P (2011) Przyczyny nieefektywnego planowania strategicznego gospodarki odpadami w Polsce - przykład Województwa Wielkopolskiego (What are the reasons for the ineffective strategicplanning of waste management in Poland? The example of the Wielkopolska Province). In: Kuczera, M. (Ed.) Wpływ Młodych Naukowców Na Osia̧gniȩcia Polskiej Nauki: 210–222. |
| [22] | Official Gazette of the Republic of Poland (2010) Resolution No. 217 of the Council of Ministries of 24 December 2010 on the "National Waste Management Plan 2014" (M.P. No. 90, item 946). |
| [23] |
Rovetta A, Xiumin F, Vicentini F, et al. (2009) Early detection and evaluation of waste through sensorized containers for a collection monitoring application. Waste Mana 29: 2939–2949. doi: 10.1016/j.wasman.2009.08.016
|
| [24] | US Environmental Protection Agency (2016) Volume-to-Weight Conversion Factors. Available from: https://www.epa.gov/sites/production/files/2016-04/documents/volume_to_weight_conversion_factors_memorandum_04192016_508fnl.pdf. |
| [25] | Kaza S, Yao L, Bhada-Tata P, et al. (2018) What a Waste 2.0: A Global Snapshot of Solid Waste Management to 2050. The World Bank, Washington. |
| [26] | Grygorczuk-Petersons EH, Wiater J (2014) Sezonowa zmienność wskaźnika nagromadzenia odpadów w wybranym osiedlu Białegostok (Seasonal variation of the waste accumulations indicator in selected estate of Białystok). Inżynieria Ekologiczna 40: 82–91. |
| [27] |
Gómez G, Meneses M, Ballinas L, et al. (2009) Seasonal characterization of municipal solid waste (MSW) in the city of Chihuahua, Mexico. Waste Manag 29: 2018–2024. doi: 10.1016/j.wasman.2009.02.006
|
| [28] | Market Analysis Department (2019) Raport Urzȩdu Ochrony Konkurencji i Konsumentów – Departament Analiz Rynku: badanie rynku usług zwia̧zanych z gospodarowaniem odpadami komunalnymi w gminach miejskich w latach 2014-2019 (Report released by Office of Competition and Consumer Protection – Market Analysis Department: market research on municipal waste management in municipalities in 2014-2019) Available from: https://www.uokik.gov.pl/aktualnosci.php?news_id=15715. |
| [29] | Przewoźna P (2019) Analiza czasowo–przestrzenna ilości odpadów komunalnych powstaja̧cych w Poznaniu (Spatial–time analysis of the amount of municipal waste generated in Poznań) Ph.D. dissertation availble from: http://hdl.handle.net/10593/24608. |
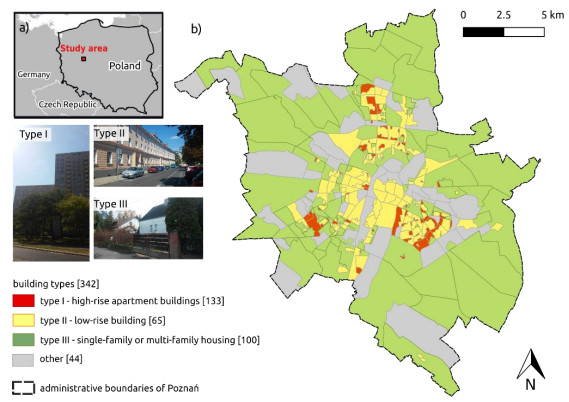
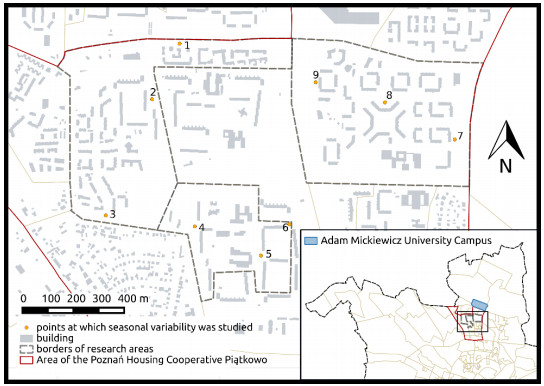
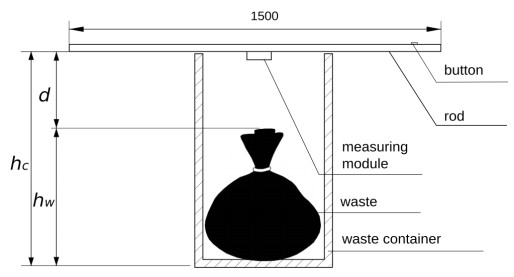
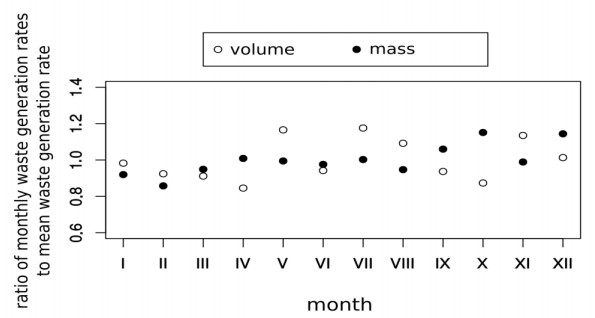
Figures(5) / Tables(1)

Patrycja Przewoźna, Piotr Jankowski, Alfred Stach. Solid waste management in urban space: the volume-weight relationship[J]. AIMS Environmental Science, 2020, 7(6): 575-588. doi: 10.3934/environsci.2020036







 DownLoad:
DownLoad: