Citation: Michael D. Caponegro, Jeremy Tetsuo Miyauchi, Stella E. Tsirka. Contributions of immune cell populations in the maintenance, progression, and therapeutic modalities of glioma[J]. AIMS Allergy and Immunology, 2018, 2(1): 24-44. doi: 10.3934/Allergy.2018.1.24

| [1] |
Wen PY, Kesari S (2008) Malignant gliomas in adults. New Engl J Med 359: 492–507. doi: 10.1056/NEJMra0708126

|
| [2] |
Brennan C, Momota H, Hambardzumyan D, et al. (2009) Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One 4: e7752. doi: 10.1371/journal.pone.0007752
|
| [3] |
Verhaak RG, Hoadley KA, Purdom E, et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110. doi: 10.1016/j.ccr.2009.12.020
|
| [4] |
Louis DN, Perry A, Reifenberger G, et al. (2016) The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol 131: 803–820. doi: 10.1007/s00401-016-1545-1
|
| [5] | Stupp R, Mason WP, Van dBMJ, et al. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med 352: 987–996. |
| [6] |
Quail DF, Joyce JA (2017) The microenvironmental landscape of brain tumors. Cancer Cell 31: 326–341. doi: 10.1016/j.ccell.2017.02.009
|
| [7] |
Lohr J, Ratliff T, Huppertz A, et al. (2011) Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res 17: 4296–4308. doi: 10.1158/1078-0432.CCR-10-2557
|
| [8] |
Kmiecik J, Poli A, Brons NH, et al. (2013) Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol 264: 71–83. doi: 10.1016/j.jneuroim.2013.08.013
|
| [9] | Yang I, Han SJ, Sughrue ME, et al. (2011a) Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: evidence of distinct immunological microenvironments that reflect tumor biology. J Neurosurg 115: 505–511. |
| [10] |
Swain SL (1995) T-cell subsets: Who does the polarizing? Curr Biol 5: 849–851. doi: 10.1016/S0960-9822(95)00170-9
|
| [11] |
Mellanby RJ, Thomas DC, Lamb J (2009) Role of regulatory T-cells in autoimmunity. Clin Sci 116: 639–469. doi: 10.1042/CS20080200
|
| [12] |
Fecci PE, Mitchell DA, Whitesides JF, et al. (2006) Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res 66: 3294–3302. doi: 10.1158/0008-5472.CAN-05-3773
|
| [13] |
Fecci PE, Ochiai H, Mitchell DA, et al. (2007) Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res 13: 2158–2167. doi: 10.1158/1078-0432.CCR-06-2070
|
| [14] |
Thomas AA, Fisher JL, Rahme GJ, et al. (2015) Regulatory T cells are not a strong predictor of survival for patients with glioblastoma. Neuro Oncol 17: 801–809. doi: 10.1093/neuonc/nou363
|
| [15] |
Han S, Zhang C, Li Q, et al. (2014) Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Brit J Cancer 110: 2560–2568. doi: 10.1038/bjc.2014.162
|
| [16] |
Sayour EJ, McLendon P, McLendon R, et al. (2015) Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol Immun 64: 419–427. doi: 10.1007/s00262-014-1651-7
|
| [17] |
Mu L, Yang C, Gao Q, et al. (2017) CD4+ and perivascular Foxp3+ T cells in glioma correlate with angiogenesis and tumor progression. Front Immunol 8: 1451. doi: 10.3389/fimmu.2017.01451
|
| [18] |
Braumuller H, Wieder T, Brenner E, et al. (2013) T-helper-1-cell cytokines drive cancer into senescence. Nature 494: 361–365. doi: 10.1038/nature11824
|
| [19] |
Hirschhorn-Cymerman D, Budhu S, Kitano S, et al. (2012) Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J Exp Med 209: 2113–2126. doi: 10.1084/jem.20120532
|
| [20] |
Perez-Diez A, Joncker NT, Choi K, et al. (2007) CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 109: 5346–5354. doi: 10.1182/blood-2006-10-051318
|
| [21] |
Murphy KA, Erickson JR, Johnson CS, et al. (2014) CD8+ T cell-independent tumor regression induced by Fc-OX40L and therapeutic vaccination in a mouse model of glioma. J Immunol 192: 224–233. doi: 10.4049/jimmunol.1301633
|
| [22] |
Saha D, Martuza RL, Rabkin SD (2017) Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell 32: 253–267. doi: 10.1016/j.ccell.2017.07.006
|
| [23] | Liu Z, Meng Q, Bartek J, et al. (2017b) Tumor-infiltrating lymphocytes (TILs) from patients with glioma. Oncoimmunology 6: e1252894. |
| [24] |
Beck BH, Kim H, O'Brien R, et al. (2015) Dynamics of circulating gammadelta T cell activity in an immunocompetent mouse model of high-grade glioma. PLoS One 10: e0122387. doi: 10.1371/journal.pone.0122387
|
| [25] |
Yang I, Tihan T, Han SJ, et al. (2010) CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J Clin Neurosci 17: 1381–1385. doi: 10.1016/j.jocn.2010.03.031
|
| [26] |
Nduom EK, Wei J, Yaghi NK, et al. (2016) PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol 18: 195–205. doi: 10.1093/neuonc/nov172
|
| [27] | Heiland DH, Haaker G, Delev D, et al. (2017) Comprehensive analysis of PD-L1 expression in glioblastoma multiforme. Oncotarget 8: 42214–42225. |
| [28] |
Reiss SN, Yerram P, Modelevsky L, et al. (2017) Retrospective review of safety and efficacy of programmed cell death-1 inhibitors in refractory high grade gliomas. J Immunother Cancer 5: 99. doi: 10.1186/s40425-017-0302-x
|
| [29] | Miyauchi JT, Tsirka SE (2017) Advances in immunotherapeutic research for glioma therapy. J Neurol, 1–16. |
| [30] |
Nakashima H, Alayo QA, Penaloza-MacMaster P, et al. (2018) Modeling tumor immunity of mouse glioblastoma by exhausted CD8+ T cells. Sci Rep 8: 208. doi: 10.1038/s41598-017-18540-2
|
| [31] |
Haanen J (2017) Converting cold into hot tumors by combining immunotherapies. Cell 170: 1055–1056. doi: 10.1016/j.cell.2017.08.031
|
| [32] |
Zeng J, See AP, Phallen J, et al. (2013) Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol 86: 343–349. doi: 10.1016/j.ijrobp.2012.12.025
|
| [33] |
Waitz R, Solomon SB, Petre EN, et al. (2012) Potent induction of tumor immunity by combining tumor cryoablation with anti-CTLA-4 therapy. Cancer Res 72: 430–439. doi: 10.1158/0008-5472.CAN-11-1782
|
| [34] |
Masson F, Calzascia T, Di BBW, et al. (2007) Brain microenvironment promotes the final functional maturation of tumor-specific effector CD8+ T cells. J Immunol 179: 845–853. doi: 10.4049/jimmunol.179.2.845
|
| [35] |
Hong JJ, Rosenberg SA, Dudley ME, et al. (2010) Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res 16: 4892–4898. doi: 10.1158/1078-0432.CCR-10-1507
|
| [36] |
Rosenberg SA, Yang JC, Sherry RM, et al. (2011) Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17: 4550–1557. doi: 10.1158/1078-0432.CCR-11-0116
|
| [37] |
Brown CE, Badie B, Barish ME, et al. (2015) Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res 21: 4062–4072. doi: 10.1158/1078-0432.CCR-15-0428
|
| [38] |
Brown CE, Alizadeh D, Starr R, et al. (2016) Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New Engl J Med 375: 2561–2569. doi: 10.1056/NEJMoa1610497
|
| [39] |
Liau LM, Prins RM, Kiertscher SM, et al. (2005) Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res 11: 5515–5525. doi: 10.1158/1078-0432.CCR-05-0464
|
| [40] |
Polyzoidis S, Ashkan K (2014) DCVax(R)-L--developed by Northwest Biotherapeutics. Hum Vacc Immunother 10: 3139–3145. doi: 10.4161/hv.29276
|
| [41] |
Flores C, Pham C, Snyder D, et al. (2015) Novel role of hematopoietic stem cells in immunologic rejection of malignant gliomas. Oncoimmunology 4: e994374. doi: 10.4161/2162402X.2014.994374
|
| [42] |
Muller I, Altherr D, Eyrich M, et al. (2016) Tumor antigen-specific T cells for immune monitoring of dendritic cell-treated glioblastoma patients. Cytotherapy 18: 1146–1161. doi: 10.1016/j.jcyt.2016.05.014
|
| [43] | Garg AD, Vandenberk L, Koks C, et al. (2016) Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med 8: 328ra27. |
| [44] |
Mitchell DA, Batich KA, Gunn MD, et al. (2015) Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 519: 366–369. doi: 10.1038/nature14320
|
| [45] |
Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19: 987–991. doi: 10.1038/nn.4338
|
| [46] |
Charles NA, Holland EC, Gilbertson R, et al. (2011) The brain tumor microenvironment. Glia 59: 1169–1180. doi: 10.1002/glia.21136
|
| [47] | Da FA, Badie B (2013) Microglia and macrophages in malignant gliomas: recent discoveries and implications for promising therapies. Clin Dev Immunol 2013: 264124. |
| [48] | Hewedi IH, Radwan NA, Shash LS, et al. (2013) Perspectives on the immunologic microenvironment of astrocytomas. Cancer Manag Res 5: 293–299. |
| [49] |
Held-Feindt J, Hattermann K, Muerkoster SS, et al. (2010) CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp Cell Res 316: 1553–1566. doi: 10.1016/j.yexcr.2010.02.018
|
| [50] | Nishie A, Ono M, Shono T, et al. (1999) Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin Cancer Res 5: 1107–1113. |
| [51] | Sorensen MD, Dahlrot RH, Boldt HB, et al. (2017) Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropath Appl Neuro 44: 185–206. |
| [52] | Okada M, Saio M, Kito Y, et al. (2009) Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. Int J Oncol 34: 1621–1627. |
| [53] |
Wang Q, Hu B, Hu X, et al. (2017) Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32: 42–56. doi: 10.1016/j.ccell.2017.06.003
|
| [54] |
Bloch O, Crane CA, Kaur R, et al. (2013) Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res 19: 3165–3175. doi: 10.1158/1078-0432.CCR-12-3314
|
| [55] |
Gieryng A, Pszczolkowska D, Bocian K, et al. (2017) Immune microenvironment of experimental rat C6 gliomas resembles human glioblastomas. Sci Rep 7: 17556. doi: 10.1038/s41598-017-17752-w
|
| [56] | Xu S, Wei J, Wang F, et al. (2014) Effect of miR-142-3p on the M2 macrophage and therapeutic efficacy against murine glioblastoma. J Natl Cancer Inst 106. |
| [57] | McFarland BC, Marks MP, Rowse AL, et al. (2016) Loss of SOCS3 in myeloid cells prolongs survival in a syngeneic model of glioma. Oncotarget 7: 20621–20635. |
| [58] |
Pyonteck SM, Akkari L, Schuhmacher AJ, et al. (2013) CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19: 1264–1272. doi: 10.1038/nm.3337
|
| [59] |
Yan D, Kowal J, Akkari L, et al. (2017) Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 36: 6049–6058. doi: 10.1038/onc.2017.261
|
| [60] |
Colegio OR, Chu NQ, Szabo AL, et al. (2014) Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513: 559–563. doi: 10.1038/nature13490
|
| [61] |
Zhang I, Alizadeh D, Liang J, et al. (2016) Characterization of arginase expression in glioma-associated microglia and macrophages. PLoS One 11: e0165118. doi: 10.1371/journal.pone.0165118
|
| [62] | Lyons YA, Pradeep S, Wu SY, et al. (2017) Macrophage depletion through colony stimulating factor 1 receptor pathway blockade overcomes adaptive resistance to anti-VEGF therapy. Oncotarget 8: 96496–96505. |
| [63] |
Nakayama T, Kurobe H, Sugasawa N, et al. (2013) Role of macrophage-derived hypoxia-inducible factor (HIF)-1alpha as a mediator of vascular remodelling. Cardiovasc Res 99: 705–715. doi: 10.1093/cvr/cvt146
|
| [64] |
Jetten N, Verbruggen S, Gijbels MJ, et al. (2014) Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 17: 109–118. doi: 10.1007/s10456-013-9381-6
|
| [65] |
Chang AL, Miska J, Wainwright DA, et al. (2016) CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory t cells and myeloid-derived suppressor cells. Cancer Res 76: 5671–5682. doi: 10.1158/0008-5472.CAN-16-0144
|
| [66] |
Solga AC, Pong WW, Kim KY, et al. (2015) RNA sequencing of tumor-associated microglia reveals Ccl5 as a stromal chemokine critical for neurofibromatosis-1 glioma growth. Neoplasia 17: 776–788. doi: 10.1016/j.neo.2015.10.002
|
| [67] |
Nissen JC, Selwood DL, Tsirka SE (2013) Tuftsin signals through its receptor neuropilin-1 via the transforming growth factor beta pathway. J Neurochem 127: 394–402. doi: 10.1111/jnc.12404
|
| [68] | Miyauchi JT, Chen D, Choi M, et al. (2016) Ablation of neuropilin 1 from glioma-associated microglia and macrophages slows tumor progression. Oncotarget 7: 9801–9814. |
| [69] |
Koncina E, Roth L, Gonthier B, et al. (2007) Role of semaphorins during axon growth and guidance. Adv Exp Med Biol 621: 50–64. doi: 10.1007/978-0-387-76715-4_4
|
| [70] |
Glinka Y, Prud'homme GJ (2008) Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J Leukoc Biol 84: 302–10. doi: 10.1189/jlb.0208090
|
| [71] |
Hu B, Guo P, Bar-Joseph I, et al. (2007) Neuropilin-1 promotes human glioma progression through potentiating the activity of the HGF/SF autocrine pathway. Oncogene 26: 5577–5586. doi: 10.1038/sj.onc.1210348
|
| [72] | Gelfand MV, Hagan N, Tata A, et al. (2014) Neuropilin-1 functions as a VEGFR2 co-receptor to guide developmental angiogenesis independent of ligand binding. Elife 3: e03720. |
| [73] |
Nakamura F, Goshima Y (2002) Structural and functional relation of neuropilins. Adv Exp Med Biol 515: 55–69. doi: 10.1007/978-1-4615-0119-0_5
|
| [74] |
Chaudhary B, Khaled YS, Ammori BJ, et al. (2014) Neuropilin 1: function and therapeutic potential in cancer. Cancer Immunol Immun 63: 81–99. doi: 10.1007/s00262-013-1500-0
|
| [75] |
Raimondi C, Fantin A, Lampropoulou A, et al. (2014) Imatinib inhibits VEGF-independent angiogenesis by targeting neuropilin 1-dependent ABL1 activation in endothelial cells. J Exp Med 211: 1167–1183. doi: 10.1084/jem.20132330
|
| [76] | Campos-Mora M, Morales RA, Gajardo T, et al. (2013) Neuropilin-1 in transplantation tolerance. Front Immunol 4: 405. |
| [77] |
Klagsbrun M, Takashima S, Mamluk R (2002) The role of neuropilin in vascular and tumor biology. Adv Exp Med Biol 515: 33–48. doi: 10.1007/978-1-4615-0119-0_3
|
| [78] |
Zachary I, Fantin A, Herzog B, et al. (2014) Neuropilin 1 (NRP1) hypomorphism combined with defective VEGF-A binding reveals novel roles for NRP1 in developmental and pathological angiogenesis. Development 141: 556–562. doi: 10.1242/dev.103028
|
| [79] |
Campos-Mora M, Morales RA, Perez F, et al. (2015) Neuropilin-1(+) regulatory T cells promote skin allograft survival and modulate effector CD4(+) T cells phenotypic signature. Immunol Cell Biol 93: 113–119. doi: 10.1038/icb.2014.77
|
| [80] |
Solomon BD, Mueller C, Chae WJ, et al. (2011) Neuropilin-1 attenuates autoreactivity in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 108: 2040–2045. doi: 10.1073/pnas.1008721108
|
| [81] |
Delgoffe GM, Woo SR, Turnis ME, et al. (2013) Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature 501: 252–256. doi: 10.1038/nature12428
|
| [82] |
Hansen W, Hutzler M, Abel S, et al. (2012) Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J Exp Med 209: 2001–2016. doi: 10.1084/jem.20111497
|
| [83] |
Milpied P, Massot B, Renand A, et al. (2011) IL-17-producing invariant NKT cells in lymphoid organs are recent thymic emigrants identified by neuropilin-1 expression. Blood 118: 2993–3002. doi: 10.1182/blood-2011-01-329268
|
| [84] |
Sarris M, Andersen KG, Randow F, et al. (2008) Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity 28: 402–413. doi: 10.1016/j.immuni.2008.01.012
|
| [85] |
Bourbie-Vaudaine S, Blanchard N, Hivroz C, et al. (2006) Dendritic cells can turn CD4+ T lymphocytes into vascular endothelial growth factor-carrying cells by intercellular neuropilin-1 transfer. J Immunol 177: 1460–1469. doi: 10.4049/jimmunol.177.3.1460
|
| [86] |
Casazza A, Laoui D, Wenes M, et al. (2013) Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 24: 695–709. doi: 10.1016/j.ccr.2013.11.007
|
| [87] |
Dejda A, Mawambo G, Daudelin JF, et al. (2016) Neuropilin-1-expressing microglia are associated with nascent retinal vasculature yet dispensable for developmental angiogenesis. Invest Ophth Vis Sci 57: 1530–1536. doi: 10.1167/iovs.15-18598
|
| [88] | Zhang W, Lv Y, Xue Y, et al. (2016b) Co-expression modules of NF1, PTEN and sprouty enable distinction of adult diffuse gliomas according to pathway activities of receptor tyrosine kinases. Oncotarget 7: 59098–59114. |
| [89] |
Glinka Y, Stoilova S, Mohammed N, et al. (2011) Neuropilin-1 exerts co-receptor function for TGF-beta-1 on the membrane of cancer cells and enhances responses to both latent and active TGF-beta. Carcinogenesis 32: 613–621. doi: 10.1093/carcin/bgq281
|
| [90] | Miyauchi JT, Caponegro MD, Chen D, et al. (2017) Deletion of neuropilin 1 from microglia or bone marrow-derived macrophages slows glioma progression. Cancer Res 78: 685–694. |
| [91] | Osada H, Tokunaga T, Nishi M, et al. (2004) Overexpression of the neuropilin 1 (NRP1) gene correlated with poor prognosis in human glioma. Anticancer Res 24: 547–552. |
| [92] |
Chen L, Miao W, Tang X, et al. (2013) Inhibitory effect of neuropilin-1 monoclonal antibody (NRP-1 MAb) on glioma tumor in mice. J Biomed Nanotechnol 9: 551–558. doi: 10.1166/jbn.2013.1623
|
| [93] |
Formolo CA, Williams R, Gordish-Dressman H, et al. (2011) Secretome signature of invasive glioblastoma multiforme. J Proteome Res 10: 3149–3159. doi: 10.1021/pr200210w
|
| [94] | Li X, Tang T, Lu X, et al. (2011) RNA interference targeting NRP-1 inhibits human glioma cell proliferation and enhances cell apoptosis. Mol Med Rep 4: 1261–1266. |
| [95] |
Nasarre C, Roth M, Jacob L, et al. (2010) Peptide-based interference of the transmembrane domain of neuropilin-1 inhibits glioma growth in vivo. Oncogene 29: 2381–2392. doi: 10.1038/onc.2010.9
|
| [96] |
Bagci T, Wu JK, Pfannl R, et al. (2009) Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene 28: 3537–3550. doi: 10.1038/onc.2009.204
|
| [97] |
Treps L, Edmond S, Harford-Wright E, et al. (2016) Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene 35: 2615–2623. doi: 10.1038/onc.2015.317
|
| [98] | Jacob L, Sawma P, Garnier N, et al. (2016) Inhibition of PlexA1-mediated brain tumor growth and tumor-associated angiogenesis using a transmembrane domain targeting peptide. Oncotarget 7: 57851–57865. |
| [99] |
Sulpice E, Plouet J, Berge M, et al. (2008) Neuropilin-1 and neuropilin-2 act as coreceptors, potentiating proangiogenic activity. Blood 111: 2036–2045. doi: 10.1182/blood-2007-04-084269
|
| [100] |
Hamerlik P, Lathia JD, Rasmussen R, et al. (2012) Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med 209: 507–520. doi: 10.1084/jem.20111424
|
| [101] |
Gilbert MR, Dignam JJ, Armstrong TS, et al. (2014) A randomized trial of bevacizumab for newly diagnosed glioblastoma. New Engl J Med 370: 699–708. doi: 10.1056/NEJMoa1308573
|
| [102] | Frei K, Gramatzki D, Tritschler I, et al. (2015) Transforming growth factor-beta pathway activity in glioblastoma. Oncotarget 6: 5963–5977. |
| [103] |
Maxwell M, Galanopoulos T, Neville-Golden J, et al. (1992) Effect of the expression of transforming growth factor-beta 2 in primary human glioblastomas on immunosuppression and loss of immune surveillance. J Neurosurg 76: 799–804. doi: 10.3171/jns.1992.76.5.0799
|
| [104] |
Platten M, Wick W, Weller M (2001) Malignant glioma biology: role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc Res Tech 52: 401–410. doi: 10.1002/1097-0029(20010215)52:4<401::AID-JEMT1025>3.0.CO;2-C
|
| [105] |
Chen ML, Pittet MJ, Gorelik L, et al. (2005) Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci USA 102: 419–424. doi: 10.1073/pnas.0408197102
|
| [106] |
Thomas DA, Massague J (2005) TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8: 369–380. doi: 10.1016/j.ccr.2005.10.012
|
| [107] |
Wesolowska A, Kwiatkowska A, Slomnicki L, et al. (2008) Microglia-derived TGF-beta as an important regulator of glioblastoma invasion--an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene 27: 918–930. doi: 10.1038/sj.onc.1210683
|
| [108] |
Uhl M, Aulwurm S, Wischhusen J, et al. (2004) SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res 64: 7954–7961. doi: 10.1158/0008-5472.CAN-04-1013
|
| [109] |
Ueda R, Fujita M, Zhu X, et al. (2009) Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res 15: 6551–6559. doi: 10.1158/1078-0432.CCR-09-1067
|
| [110] |
Rodon J, Carducci MA, Sepulveda-Sanchez JM, et al. (2015) First-in-human dose study of the novel transforming growth factor-beta receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res 21: 553–560. doi: 10.1158/1078-0432.CCR-14-1380
|
| [111] |
Falco SD (2012) The discovery of placenta growth factor and its biological activity. Exp Mol Med 44: 1–9. doi: 10.3858/emm.2012.44.1.025
|
| [112] | Dewerchin M, Carmeliet P (2012) PlGF: a multitasking cytokine with disease-restricted activity. Csh Perspect Med 2: 143–152. |
| [113] |
Lassen U, Chinot OL, McBain C, et al. (2015) Phase 1 dose-escalation study of the antiplacental growth factor monoclonal antibody RO5323441 combined with bevacizumab in patients with recurrent glioblastoma. Neuro Oncol 17: 1007–1015. doi: 10.1093/neuonc/nov019
|
| [114] |
Snuderl M, Batista A, Kirkpatrick ND, et al. (2013) Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell 152: 1065–1076. doi: 10.1016/j.cell.2013.01.036
|
| [115] |
Guo Y, Wang X, Tian X, et al. (2012) Tumor-derived hepatocyte growth factor is associated with poor prognosis of patients with glioma and influences the chemosensitivity of glioma cell line to cisplatin in vitro. World J Surg Oncol 10: 1–11. doi: 10.1186/1477-7819-10-1
|
| [116] |
Wen PY, Schiff D, Cloughesy TF, et al. (2011) A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol 13: 437–446. doi: 10.1093/neuonc/noq198
|
| [117] | De Groot JF, Prados M, Urquhart T, et al. (2009) A phase II study of XL184 in patients (pts) with progressive glioblastoma multiforme (GBM) in first or second relapse. J Clin Oncol 27: 2047. |
| [118] |
Schiff D, Desjardins A, Cloughesy T, et al. (2016) Phase 1 dose escalation trial of the safety and pharmacokinetics of cabozantinib concurrent with temozolomide and radiotherapy or temozolomide after radiotherapy in newly diagnosed patients with high-grade gliomas. Cancer 122: 582–587. doi: 10.1002/cncr.29798
|
| [119] |
Wu HB, Wang Z, Wang QS, et al. (2015) Use of labelled tLyP-1 as a novel ligand targeting the NRP receptor to image glioma. PLoS One 10: e0137676. doi: 10.1371/journal.pone.0137676
|
| [120] |
Ying M, Shen Q, Zhan C, et al. (2016) A stabilized peptide ligand for multifunctional glioma targeted drug delivery. J Control Release 243: 86–98. doi: 10.1016/j.jconrel.2016.09.035
|
| [121] |
Ying M, Zhan C, Wang S, et al. (2016) Liposome-based systemic glioma-targeted drug delivery enabled by all-d peptides. ACS Appl Mater Inter 8: 29977–29985. doi: 10.1021/acsami.6b10146
|
| [122] |
Liu C, Yao S, Li X, et al. (2017) iRGD-mediated core-shell nanoparticles loading carmustine and O6-benzylguanine for glioma therapy. J Drug Target 25: 235–246. doi: 10.1080/1061186X.2016.1238091
|
| [123] |
Liu Y, Mei L, Xu C, et al. (2016) Dual receptor recognizing cell penetrating peptide for selective targeting, efficient intratumoral diffusion and synthesized anti-glioma therapy. Theranostics 6: 177–191. doi: 10.7150/thno.13532
|
| [124] |
Bechet D, Auger F, Couleaud P, et al. (2015) Multifunctional ultrasmall nanoplatforms for vascular-targeted interstitial photodynamic therapy of brain tumors guided by real-time MRI. Nanomed-Nanotechnol 11: 657–670. doi: 10.1016/j.nano.2014.12.007
|
| [125] |
Patnaik A, Lorusso PM, Messersmith WA, et al. (2014) A Phase Ib study evaluating MNRP1685A, a fully human anti-NRP1 monoclonal antibody, in combination with bevacizumab and paclitaxel in patients with advanced solid tumors. Cancer Chemoth Pharm 73: 951–960. doi: 10.1007/s00280-014-2426-8
|
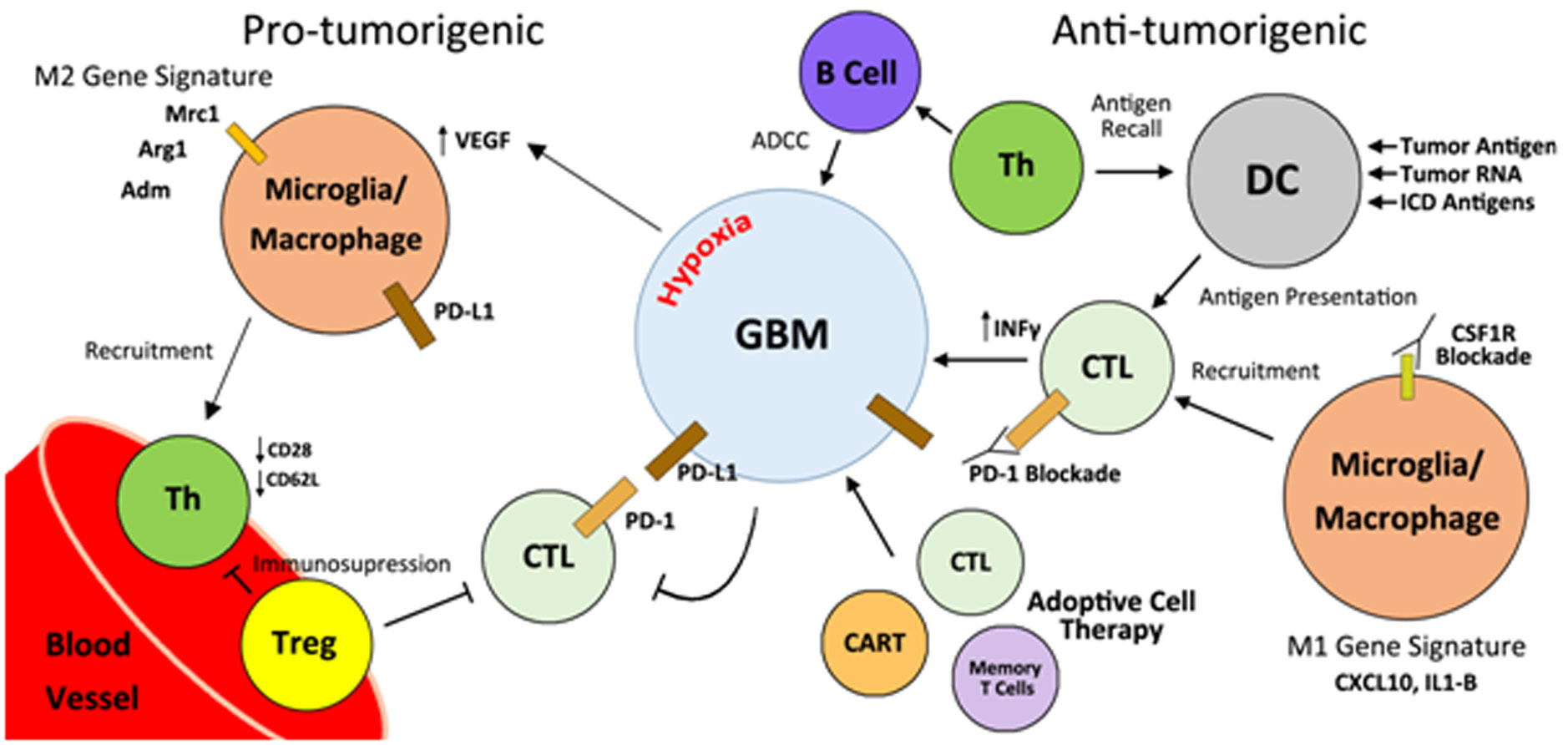
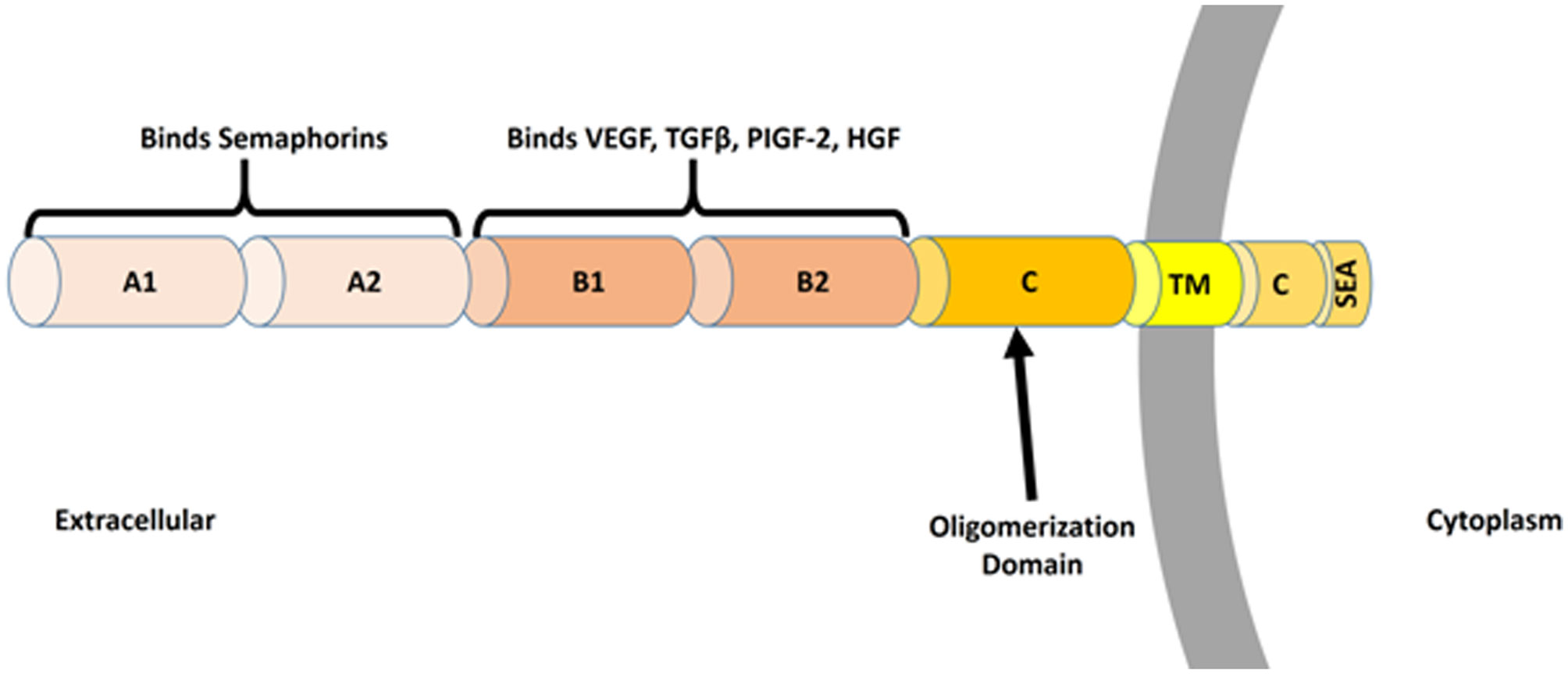
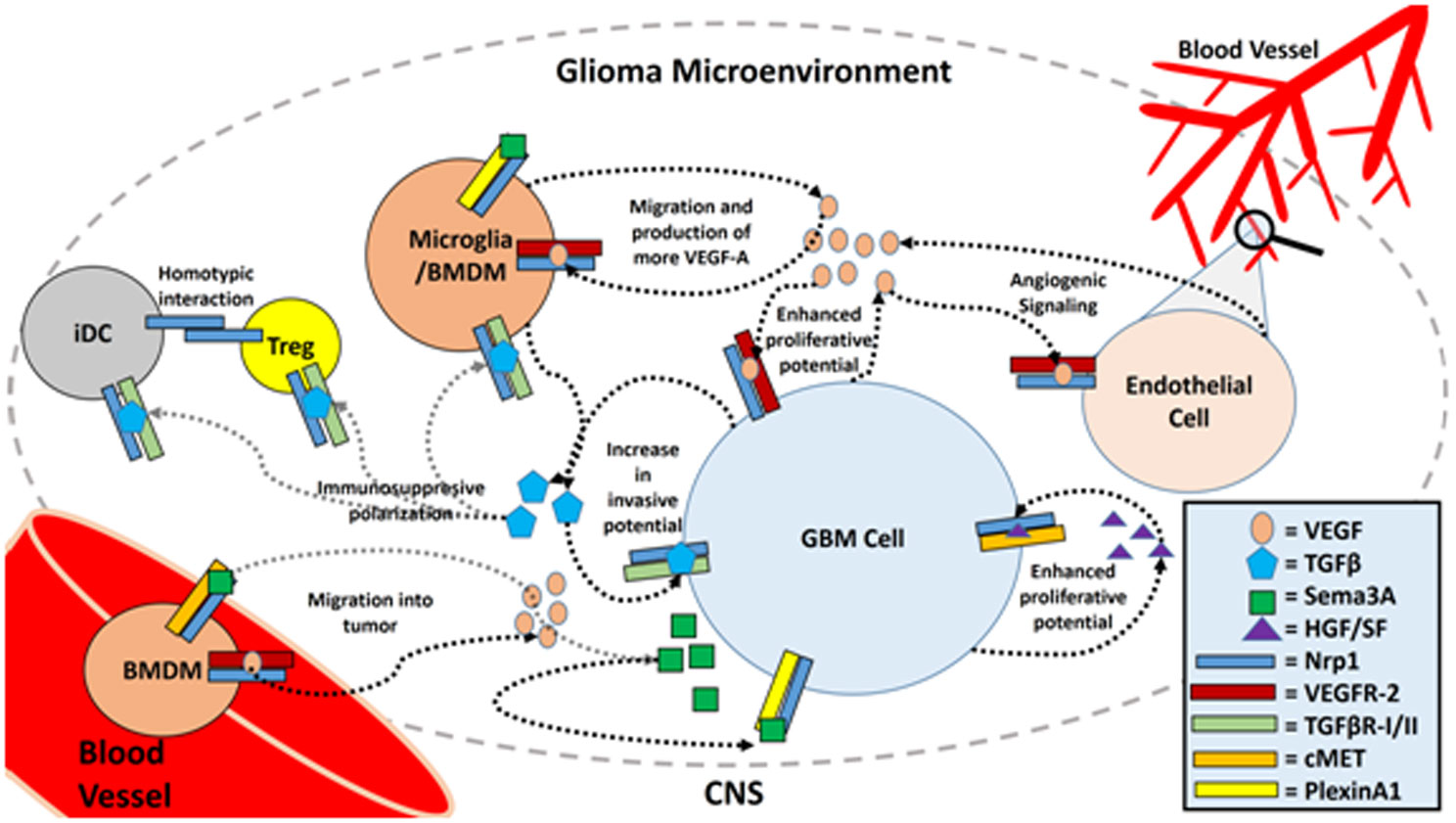
Figures(3)

Michael D. Caponegro, Jeremy Tetsuo Miyauchi, Stella E. Tsirka. Contributions of immune cell populations in the maintenance, progression, and therapeutic modalities of glioma[J]. AIMS Allergy and Immunology, 2018, 2(1): 24-44. doi: 10.3934/Allergy.2018.1.24







 DownLoad:
DownLoad: