Citation: Laura Serna. Crosstalk among hormones and signaling networks during stomatal development in Arabidopsis hypocotyls[J]. AIMS Molecular Science, 2016, 3(4): 550-559. doi: 10.3934/molsci.2016.4.550

| [1] |
Gendreau E, Traas J, Desnos T, et al. (1997) Cellular basis of hypocotyl growth in Arabidopsis thaliana. Plant Physiol 114: 295-305. doi: 10.1104/pp.114.1.295

|
| [2] |
Hung C-Y, Lin Y, Zhang M, et al. (1998) A common position-dependent mechanism controls cell-type patterning and GLABRA2 regulation in the root and hypocotyl epidermis of Arabidopsis. Plant Physiol 117: 73-84. doi: 10.1104/pp.117.1.73
|
| [3] |
Berger F, Linstead P, Dolan L, et al. (1998) Stomata patterning on the hypocotyl of Arabidopsis thaliana is controlled by genes involved in the control of root epidermis patterning. Dev Biol 194: 226-234. doi: 10.1006/dbio.1997.8836
|
| [4] |
Saibo NJ, Vriezen WH, Beemster GT, et al. (2003) Growth and stomata development of Arabidopsis hypocotyls are controlled by gibberellins and modulated by ethylene and auxins . Plant J 33: 989-1000. doi: 10.1046/j.1365-313X.2003.01684.x
|
| [5] |
Fuentes S, Cañamero RC, Serna L (2012) Relationship between brassinosteroids and genes controlling stomatal production in the Arabidopsis hypocotyl. Int J Dev Biol 56: 675-680. doi: 10.1387/ijdb.120029ls
|
| [6] | GudesblatGE,Schneider-PizońJ,BettiC,et al. (2012)SPEECHLESS integrates brassinosteroid and stomata signaling pathways. Nat Cell Biol 14: 548-554. |
| [7] |
Bhave NS, Veley KM, Nadeau JA, et al. (2009) TOO MANY MOUTHS promotes cell fate progression in stomatal development of Arabidopsis stems. Planta 229: 357-367. doi: 10.1007/s00425-008-0835-9
|
| [8] |
Lee MM, Schiefelbein J (1999) WEREWOLF, a MYB-related protein in Arabidopis, is a position-dependent regulator of epidermal cell patterning. Cell 99: 473-483. doi: 10.1016/S0092-8674(00)81536-6
|
| [9] |
Bernhardt C, Zhao M, Gonzalez A, et al. (2005) The bHLH genes GL3 and EGL3 participate in an intercellular regulatory circuit that controls cell patterning in the Arabidopsis root epidermis. Development 132: 291-298. doi: 10.1242/dev.01565
|
| [10] |
Walker AR, Davison PA, Bolognesi-Winfield AC, et al. (1999) The TRANSPARENT TESTA GLABRA1 locus, which regulates trichome differentiation and anthocyanin biosynthesis in Arabidopsis, encodes a WD40 repeat protein. Plant Cell 11: 1337-1349. doi: 10.1105/tpc.11.7.1337
|
| [11] | Payne CT, Zhang F, Lloyd AM (2000) GL3 encodes a bHLH protein that regulates trichome development in Arabidopsis through interaction with GL1 and TTG1. Genetics 156: 1349-1362. |
| [12] |
Zhang F, Gonzalez A, Zhao M, et al. (2003) A network of redundant bHLH proteins functions in all TTG1-dependent pathways of Arabidopsis. Development 130: 4859-4869. doi: 10.1242/dev.00681
|
| [13] | Bernhardt C, Lee MM, Gonzalez A, et al. (2003) The bHLH genes GLABRA3. GL3, and ENHANCER OF GLABRA3. EGL3, specify epidermal cell fate in the Arabidopsis root. Development 130: 6431-6439. |
| [14] | Larkin JC, Brown ML, Schiefelbein J (2003) How cells know what they want to be when they grow up? Lessons from epidermal patterning in Arabidopsis. Ann Rev Plant Biol 54: 403-430. |
| [15] |
Schiefelbein J (2003) Cell-fate specification in the epidermis: a common patterning mechanism in the root and shoot. Curr Opin Plant Biol 6: 74-78. doi: 10.1016/S136952660200002X
|
| [16] | Serna L (2004a) Epidermal cell patterning and differentiation throughout the apical-basal axis of the seedling. J Exp Bot 56: 1983-1989. |
| [17] | Serna L (2004b) A network of interacting factors triggering different cell fates. Plant Cell 16: 2258-2263. |
| [18] |
Rerie WG, Feldmann KA, Marks MD (1994) The GLABRA2 gene encodes a homeodomain protein required for normal trichome development in Arabidopsis. Genes Dev 8: 1388-1399. doi: 10.1101/gad.8.12.1388
|
| [19] |
Di Cristina MD, Sessa G, Dolan L, et al. (1996) The Arabidopsis Athb-10 (GLABRA2) is an HD-Zip protein required for regulation of root hair development. Plant J 10: 393-402. doi: 10.1046/j.1365-313X.1996.10030393.x
|
| [20] |
Serna L (2008) CAPRICE positively regulates stomatal formation in the Arabidopsis hypocotyl. Plant Signal Behav 3: 1-6. doi: 10.4161/psb.3.1.5279
|
| [21] |
Esch JJ, Chen M, Sanders M, et al. (2003) A contradictory GLABRA3 allele helps define gene interactions controlling trichome development in Arabidopsis. Development 130: 5885-5894. doi: 10.1242/dev.00812
|
| [22] |
Kirik V, Simon M, Huelskamp M, et al. (2004) The ENHANCER OF TRY AND CPC1 gene acts redundantly with TRIPTYCHON and CAPRICE in trichome and root hair cell patterning in Arabidopsis. Dev Biol 268: 506-513. doi: 10.1016/j.ydbio.2003.12.037
|
| [23] | RademacherW (2000)Growth retardants: effects on gibberellin biosynthesis and other metabolic pathways. Ann RevPlant PhysiolPlant Mol Biol 51:501-531. |
| [24] | Behringer C,Schwechheimer C (2015) B-GATA transcription factors - insights into their structure, regulation, and role in plant development. Front Plant Sci 6: 90. |
| [25] |
Klermund C,Ranftl QL,Diener J,et al. (2016) LLM-domain B-GATA transcription factors promote stomatal development downstream of light signaling pathways in Arabidopsis thaliana hypocotyls. Plant Cell 28: 646-660. doi: 10.1105/tpc.15.00783
|
| [26] |
Bai M-Y, Shang J-X, Oh E, et al. (2012) Brassinosteroid, gibberellin and phytochrome impinge on a common transcription module in Arabidopsis. Nat Cell Biol 14: 810-817. doi: 10.1038/ncb2546
|
| [27] |
Gallego-Bartolomé J,Minguet EG,Grau-Enguix F, et al. (2012) Molecular mechanism for the interaction between gibberellin and brassinosteroid signaling pathways in Arabidopsis. Proc Natl Acad Sci U S A 109: 13446-13451. doi: 10.1073/pnas.1119992109
|
| [28] | Li QF,Wang C,Jiang L,et al. (2012) An interaction between BZR1 and DELLAs mediates direct signaling crosstalk between brassinosteroids and gibberellins in Arabidopsis. Sci Signal 5: ra72. |
| [29] |
Serna L (2013) What causes opposing actions of brassinosteroids on stomatal development? Plant Physiol 162: 3-8. doi: 10.1104/pp.112.213058
|
| [30] |
Kim T-W, Michniewicz M, Bergmann DC, et al. (2012). Brassinosteroid regulates stomatal development by GSK3-mediated inhibition of a MAPK pathway. Nature 482: 419-422. doi: 10.1038/nature10794
|
| [31] |
Khan M,Rozhon W,Bigeard J, et al. (2013). Brassinosteroid-regulated GSK3/Shaggy-like kinases phosphorylate mitogen-activated protein (MAP) kinase kinases, which control stomata development in Arabidopsis thaliana . J Biol Chem 288: 7519-7527. doi: 10.1074/jbc.M112.384453
|
| [32] |
Balcerowicz M,Ranjan A,Rupprecht L,et al. (2014) Auxin represses stomatal development in dark-grown seedlings via Aux/IAA proteins. Development 141: 3165-3176. doi: 10.1242/dev.109181
|
| [33] |
Balcerowicz M,Hoecker U (2014) Auxin - a novel regulator of stomata differentiation. Trends Plant Sci 19: 747-749. doi: 10.1016/j.tplants.2014.10.006
|
| [34] |
Kuppusamy KT,Chen AY,Nemhauser JL (2009) Steroids are required for epidermal cell fate establishment in Arabidopsis roots. Proc Natl Acad Sci U S A 106: 8073-8076. doi: 10.1073/pnas.0811633106
|
| [35] |
Perazza D, Vachon G,Herzog M (1998) Gibberellins Promote Trichome Formation by Up-Regulating GLABROUS1 in Arabidopsis. Plant Physiol 117: 375-383. doi: 10.1104/pp.117.2.375
|
| [36] |
Nadeau JA, Sack FD (2002) Control of stomatal distribution on the Arabidopsis leaf surface. Science 296: 1697-1700. doi: 10.1126/science.1069596
|
| [37] |
Wang M,Yang K,Le J (2015) Organ-specific effects of brassinosteroids on stomatal production coordinate with the action of Too Many Mouths. J Integr Plant Biol 57: 247-255. doi: 10.1111/jipb.12285
|
| [38] |
MacAlister CA, Ohashi-Ito K, Bergmann DC (2007) Transcription factor control of asymmetric cell divisions that establish the stomatal lineage. Nature 445: 537-540. doi: 10.1038/nature05491
|
| [39] | Pillitteri LJ, Sloan DB, Bogenschutz NL, et al. (2007) Termination of asymmetric cell division and differentiation of stomata. Nature 445, 501-505. |
| [40] |
Robinson S, Barbier de Reuille P, Chan J, et al. (2011) Generation of spatial patterns through cell polarity switching. Science 333: 1436-1440. doi: 10.1126/science.1202185
|
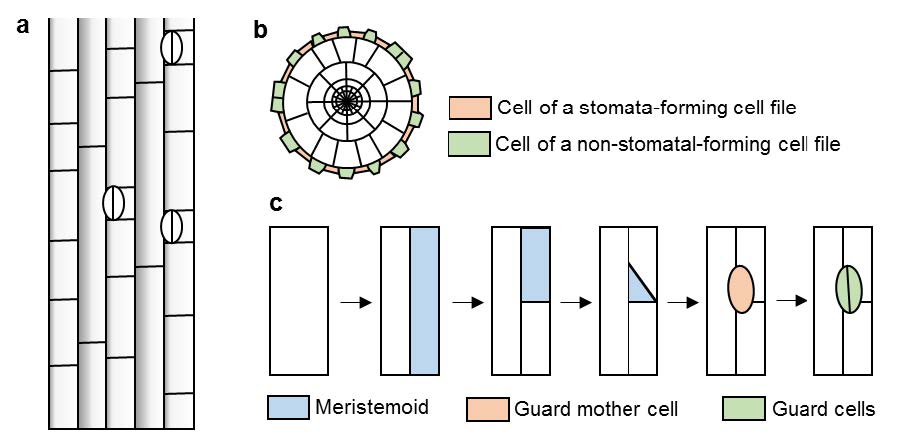
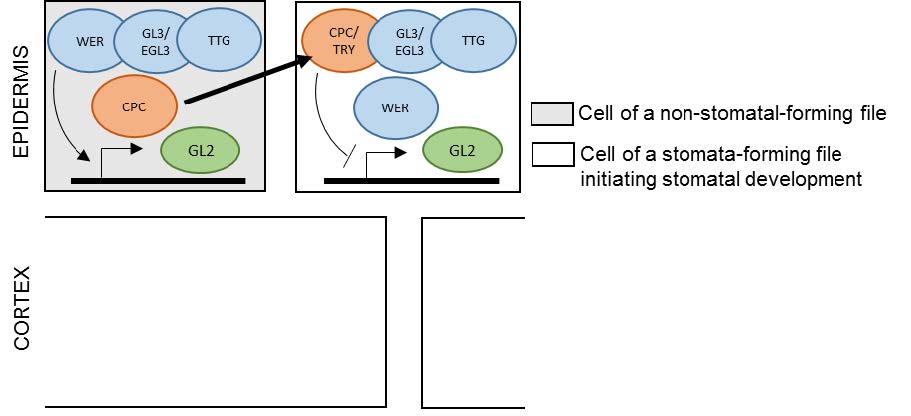
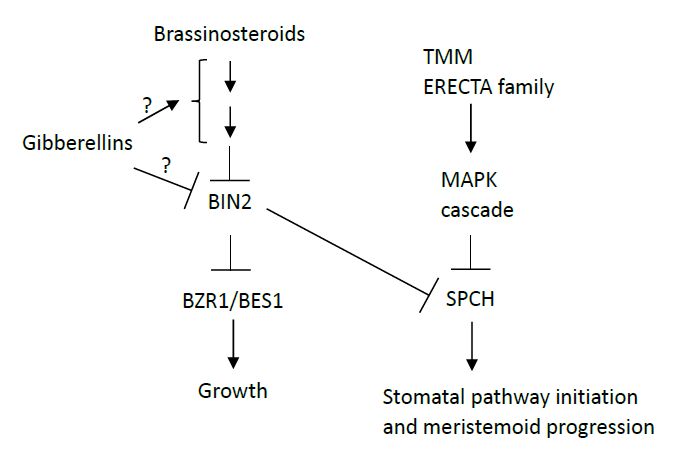
Figures(3)

Laura Serna. Crosstalk among hormones and signaling networks during stomatal development in Arabidopsis hypocotyls[J]. AIMS Molecular Science, 2016, 3(4): 550-559. doi: 10.3934/molsci.2016.4.550







 DownLoad:
DownLoad: