Citation: Valentina Contrò, John R. Basile, Patrizia Proia. Sex steroid hormone receptors, their ligands, and nuclear and non-nuclear pathways[J]. AIMS Molecular Science, 2015, 2(3): 294-310. doi: 10.3934/molsci.2015.3.294

| [1] |
Pietras RJ, Szego CM (1977) Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature 265: 69-72. doi: 10.1038/265069a0

|
| [2] | Pietras RJ, Szego CM (1980) Partial purification and characterization of oestrogen receptors in subfractions of hepatocyte plasma membranes. Biochem J 191: 743-760. |
| [3] | Tremblay GB, Tremblay A, Copeland NG, et al. (1997) Cloning, chromosomal localization and functional analysis of the murine estrogen receptor beta. Mol Endocrinol 11: 353-365. |
| [4] |
Leitman DC, Paruthiyil S, Yuan C, et al. (2012) Tissue specific regulation of genes by estrogen receptors Sem. Reprod Med 30: 14-22 doi: 10.1055/s-0031-1299593
|
| [5] |
Meneses-Morales I, Tecalco-Cruz AC, Barrios-García T, et al. (2014) SIP1/NHERF2 enhance estrogen receptor alpha transactivation in breast cancer cells. Nucleic Acids Res 42: 6885-6900. doi: 10.1093/nar/gku311
|
| [6] | Wright PK, May FEB, Darby S, et al. (2009) Estrogen regulates vesicle trafficking gene expression in EFF-3, EFM-19 and MCF-7 breast cancer cells. Int J Clin Exp Pathol 2: 463-475. |
| [7] |
Manavathi B, Dey O, Gajulapalli VNR, et al. (2013) Derailed estrogen signaling and breast cancer: an authentic couple. Endocr Rev 34: 1-32 doi: 10.1210/er.2011-1057
|
| [8] |
Mani SK, Mermelstein PG, Tetel MJ, et al. (2012) Convergence of Multiple Mechanisms of Steroid Hormone Action. Horm Metab Res 44: 569-576. doi: 10.1055/s-0032-1306343
|
| [9] | Zhao C, Dahlman-Wright K, Gustafsson JA (2008) Estrogen receptor beta: an overview and update. Nucl Recept Signal 6: e003. |
| [10] |
Mooradian AD, Morley JE, Korenman SC (1987) Biological actions of androgens. Endocr Rev 8: 1-28 doi: 10.1210/edrv-8-1-1
|
| [11] |
Picard D (2006) Chaperoning steroid hormone action. Trends Endocrinol Metab 17: 229-235. doi: 10.1016/j.tem.2006.06.003
|
| [12] |
Kicman AT (2008) Pharmacology of anabolic steroids. Brit J Pharmacol 154: 502-521 doi: 10.1038/bjp.2008.165
|
| [13] | Falkenstein E, Tillmann HC, Christ M, et al. (2000) Multiple action of steroid hormones-a focus on rapid, nongenomic effects. Pharmacol Rev 52: 513-56. |
| [14] | Cato AC, Nestl A, Mink S (2002) Rapid action of steroid receptors in cellular signaling pathways. Sci STKE 138: re9 |
| [15] | Liao RS, Ma S, Miao L, et al. (2013) Androgen receptor-mediated non-genomic regulation of prostate cancer cell proliferation. Transl Androl Urol 2: 187-196 |
| [16] | Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1: 31-39. |
| [17] |
Pedram A, Razandi M, Sainson RC, et al. (2007) A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem 282: 22278-22288. doi: 10.1074/jbc.M611877200
|
| [18] | Kousteni S, Bellido T, Plotkin LI, et al. (2001) Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity Cell 104: 719-730. |
| [19] | Y Gong, L J Blok, J E Perry, et al. (1995) Calcium regulation of androgen receptor expression in the human prostate cancer cell line LNCaP. Endocrinology 136: 2172-2178 |
| [20] | Trabert B, Wentzensen N, Yang HP, et al. (2013) Is estrogen plus progestin menopausal hormone therapy safe with respect to endometrial cancer risk? Int J Cancer 15: 417-26. |
| [21] | Westley RL, May FEB (2013) A twenty-first century cancer epidemic caused by obesity: the involvement of insulin, diabetes, and insulin-like growth factors. Int J Endocrinol 2013: 632461. |
| [22] |
Mady EA, Ramadan EE, Ossman AA (2000) Sex steroid hormones in serum and tissue of benign and malignant breast tumor patients. Dis Markers 16: 151-157. doi: 10.1155/2000/305940
|
| [23] |
Allen NE, Beral V, Casabonne D, et al. (2009) Moderate alcohol intake and cancer incidence in women. J Natl Cancer Inst 101: 296-305. doi: 10.1093/jnci/djn514
|
| [24] |
McCullough LE, Eng SM, Bradshaw PT, et al. (2012) Fat or fit: the joint effects of physical activity, weight gain, and body size on breast cancer risk. Cancer 118: 4860-4868. doi: 10.1002/cncr.27433
|
| [25] | Siddappa K (2002) Cutaneous and mucosal pain syndromes. Indian J Dermatol Venereol Leprol 68: 123-130 |
| [26] |
Piérard-Franchimont C, Piérard GE (2002) Postmenopausal aging of the sebaceous follicle: a comparison between women receiving hormone replacement therapy or not. Dermatology 204: 17-22. doi: 10.1159/000051804
|
| [27] | Klein-Nulend J, van Oers RF, Bakker AD, et al. (2014) Bone cell mechanosensitivity, estrogen deficiency, and osteoporosis. J Biomech S0021-9290: 00661-7 |
| [28] | Shen M, Kumar SP, Shi H (2014) Estradiol regulates insulin signaling and inflammation in adipose tissue. Horm Mol Biol Clin Investig 17: 99-107. |
| [29] |
Hammes SR, Levin ER (2011) Minireview: Recent Advances in Extranuclear Steroid Receptor Actions. Endocrinology152: 4489-4495. doi: 10.1210/en.2011-1470
|
| [30] |
Collins P, Webb C (1999) Estrogen hits the surface. Nat Med 5: 1130-1131. doi: 10.1038/13453
|
| [31] | Kajiwara M1, Kuraku S, Kurokawa T, et al. (2006). Tissue preferential expression of estrogen receptor gene in the marine snail, Thais clavigera. Gen Comp Endocrinol 148: 315-326 |
| [32] |
Koike S, Sakai M, Muramatsu M (1987) Molecular cloning and characterization of rat estrogen receptor cDNA. Nucleic Acids Res 15: 2499-2513. doi: 10.1093/nar/15.6.2499
|
| [33] |
Kuiper GG, Enmark E, Pelto-Huikko M, et al. (1996) Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A 93: 5925-5930. doi: 10.1073/pnas.93.12.5925
|
| [34] | Giguere V (2008) Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev 13: 677-696. |
| [35] |
Levin ER (2012) Elusive extranuclear estrogen receptors in breast cancer. Clin Cancer Res 18: 6-8 doi: 10.1158/1078-0432.CCR-11-2547
|
| [36] |
Pedram A, Razandi M, Wallace DC, et al. (2006) Functional estrogen receptors in the mitochondria of breast cancer cells. Mol Biol Cell 17: 2125-2137. doi: 10.1091/mbc.E05-11-1013
|
| [37] |
Giguere V (2002) To ERR in the estrogen pathway. Trends Endocrinol Metab 13: 220-225. doi: 10.1016/S1043-2760(02)00592-1
|
| [38] | Sladek R, Bader J-A, Giguere V (1997) The orphan nuclear receptor estrogen-related receptor alpha is a transcriptional regulator of the human medium-chain acyl coenzyme A dehydrogenase gene. Mol Cell Biol 17: 5400-5409. |
| [39] | Deblois G, Hall JA, Perry MC, et al. (2009) Genome-wide identification of direct target genes implicates estrogen-related receptor alpha as a determinant of breast cancer heterogeneity. Cancer Res 13: 6149-6157. |
| [40] |
Hall JM, Couse JF, Korach KS (2001) The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem 276: 36869-36872. doi: 10.1074/jbc.R100029200
|
| [41] |
Bernatchez G, Giroux V, Lassalle T, et al. (2013) ERRα metabolic nuclear receptor controls growth of colon cancer cells. Carcinogenesis 34: 2253-2261. doi: 10.1093/carcin/bgt180
|
| [42] | Chen P, Wang H, Duan Z, et al. (2014) Estrogen-related receptor alpha confers methotrexate resistance via attenuation of reactive oxygen species production and P53 mediated apoptosis in osteosarcoma cells. Biomed Res Int 2014: 616025. |
| [43] |
Herzog B, Cardenas J, Hall RK, et al. (2006) Estrogen-related receptor alpha is a repressor of phosphoenolpyruvate carboxykinase gene transcription. J Biol Chem 281: 99-106. doi: 10.1074/jbc.M509276200
|
| [44] |
Klinge CM (2001) Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res 29: 2905-2919. doi: 10.1093/nar/29.14.2905
|
| [45] | May FEB (2014) Novel drugs that target the estrogen-related receptor alpha: their therapeutic potential in breast cancer. Cancer Manag Res 6: 225-252 |
| [46] | Byerly MS, Al Salayta M, Swanson RD, et al. (2013) Estrogen-related receptor beta deletion modulates whole-body energy balance via estrogen-related receptor gamma and attenuates neuropeptide Y gene expression. Eur J Neurosci 13: 1033-1047. |
| [47] |
Misra J, Chanda D, Kim DK, et al. (2014) Orphan nuclear receptor Errγ induces C-reactive protein gene expression through induction of ER-bound Bzip transmembrane transcription factor CREBH. PLoS One 9: e86342. doi: 10.1371/journal.pone.0086342
|
| [48] | Wang Z, Li P, Zhang Q, et al. (2015) Interleukin-1β regulates the expression of glucocorticoid receptor isoforms in nasal polyps in vitro via p38 MAPK and JNK signal transduction pathways. J Inflamm (Lond) 12: 3. |
| [49] |
Revankar CM, Cimino DF, Sklar LA, et al. (2005) A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307: 1625-1630. doi: 10.1126/science.1106943
|
| [50] |
Chevalier N, Paul-Bellon R, Camparo P, et al. (2014) Genetic variants of GPER/GPR30, a novel estrogen-related G protein receptor, are associated with human seminoma. Int J Mol Sci 15: 1574-1589. doi: 10.3390/ijms15011574
|
| [51] |
Shahani S, Braga-Basaria M, Maggio M, et al. (2009) Androgens and erythropoiesis: past and present. J Endocrinol Invest 32: 704-716 doi: 10.1007/BF03345745
|
| [52] |
Furuya K, Yamamoto N, Ohyabu Y, et al. (2013) Mechanism of the tissue-specific action of the selective androgen receptor modulator S-101479. Biol Pharm Bull 36: 442-451. doi: 10.1248/bpb.b12-00885
|
| [53] | Roy AK, Lavrovsky Y, Song CS, et al. (1999) Regulation of androgen action. Vitam Horm 55: 309-352 |
| [54] | Fortunati N (1999) Sex hormone-binding globulin: not only a transport protein. What news around the corner? J Endocrinol Invest 22: 223-234 |
| [55] |
Rosner W, Hryb DJ, Khan MS, et al. (1999) Sex hormone-binding globulin mediates steroid hormone signal transduction at the plasma membrane. J Steroid Biochem Mol Biol 69: 481-485 doi: 10.1016/S0960-0760(99)00070-9
|
| [56] | Fortunati N, Catalano MG, Boccuzzi G, et al. (2010) Sex Hormone-Binding Globulin (SHBG), estradiol and breast cancer. Mol Cell Endocrinol 316: 86-92 |
| [57] |
Sá EQ, Sá FC, Oliveira KC, et al. (2014) Association between sex hormone- binding globulin (SHBG) and metabolic syndrome among men. Sao Paulo Med J 132: 111-115 doi: 10.1590/1516-3180.2014.1322666
|
| [58] |
Herbert Z, Göthe S, Caldwell JD, et al. (2005) Identification of sex hormone-binding globulin in the human hypothalamus. Neuroendocrinology 81: 287-293. doi: 10.1159/000088170
|
| [59] |
Misao R, Nakanishi Y, Fujimoto J, et al. (1995) Expression of sex hormone-binding globulin mRNA in uterine leiomyoma, myometrium and endometrium of human subjects. Gynecol Endocrinol 9: 317-323 doi: 10.3109/09513599509160466
|
| [60] | Selva DM, Hammond GL (2006) Human sex hormone-binding globulin is expressed in testicular germ cells and not in sertoli cells. Horm Metab Res 38: 230-235 |
| [61] |
Heinlein CA, Chang C (2002) The roles of androgen receptors and androgen-binding proteins in nongenomic androgen actions. Mol Endoc 16: 2181-2187 doi: 10.1210/me.2002-0070
|
| [62] |
Inoue K, Yamasaki S, Fushiki T, et al. (1994) Androgen receptor antagonist suppresses exercise-induced hypertrophy of skeletal muscle. Eur J Appl Physiol Occup Physiol 69: 88-91. doi: 10.1007/BF00867933
|
| [63] | Bamman MM, Shipp JR, Jiang J, et al. (2001) Mechanical load increases muscle IGF-I and androgen receptor mRNA concentrations in humans. Am J Physiol Endocrinol Metab 280: 383-390. |
| [64] |
Kadi F (2008) Testosterone and human skeletal muscle. Brit J Pharmacol 154: 522-528. doi: 10.1038/bjp.2008.118
|
| [65] |
Sinha-Hikim I, Taylor WE, Gonzalez-Cadavid NF, et al. (2004) Androgen receptor in human skeletal muscle and cultured muscle satellite cells: up-regulation by androgen treatment. J Clin Endocrinol Metab 89: 5245-5255. doi: 10.1210/jc.2004-0084
|
| [66] |
Hoffman JR1, Kraemer WJ, Bhasin S, et al. (2009) Position stand on androgen and human growth hormone use. J Strength Cond Res 23: S1-59. doi: 10.1519/JSC.0b013e31819df2e6
|
| [67] | Basile J, Binmadi N, Zhou H, et al. (2013) Supraphysiological doses of performance enhancing anabolic-androgenic steroids exert direct tossic effects on neuron-like cells. Front Cell Neurosc 7: 1-10 |
| [68] |
Castoria G, D'Amato L, Ciociola A, et al. (2011) Androgen-Induced Cell Migration: Role of Androgen Receptor/Filamin A Association. PLoS ONE 6: e17218. doi: 10.1371/journal.pone.0017218
|
| [69] | Giovannelli P, Di Donato M, Cernera G, et al. (2015) The dual role of androgen receptor in mesenchymal cells. Receptor Clin Invest 2: e664. |
| [70] |
Papadopoulou N, Papakonstanti EA, Kallergi G, et al. (2009) Membrane androgen receptor activation in prostate and breast tumor cells: molecular signaling and clinical impact. IUBMB Life 61: 56-61 doi: 10.1002/iub.150
|
| [71] |
Bae YH, Mui KL, Hsu BY, et al. (2014) A FAK-Cas-Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci Signal 7: ra57. doi: 10.1126/scisignal.2004838
|
| [72] |
De Naeyer H, Bogaert V, De Spaey A, et al. (2014) Genetic variations in the androgen receptor are associated with steroid concentrations and anthropometrics but not with muscle mass in healthy young men. PLoS One 9: e86235. doi: 10.1371/journal.pone.0086235
|
| [73] |
Brinkmann AO, Faber PW, van Rooij HC, et al. (1989) The human androgen receptor: domain structure, genomic organization and regulation of expression. J Steroid Biochem 34: 307-310. doi: 10.1016/0022-4731(89)90098-8
|
| [74] |
Jänne OA, Bardin CW (1984) Androgen and antiandrogen receptor binding. Annu Rev Physiol 46: 107-118. doi: 10.1146/annurev.ph.46.030184.000543
|
| [75] | Garay JP, Park BH (2012) Androgen receptor as a targeted therapy for breast cancer. Am J Cancer Res 2: 434-445. |
| [76] |
Narayanan R, Ahn S, Cheney MD, et al. (2014) Selective androgen receptor modulators (SARMs) negatively regulate triple-negative breast cancer growth and epithelial: mesenchymal stem cell signaling. PLoS One 9: e103202. doi: 10.1371/journal.pone.0103202
|
| [77] |
Wang C, Liu Y, Cao JM (2014) G protein-coupled receptors: extranuclear mediators for the non-genomic actions of steroids. Int J Mol Sci 15: 15412-15425. doi: 10.3390/ijms150915412
|
| [78] | Pi M, Parrill AL, Quarles LD (2010) GPRC6A mediates the non-genomic effects of steroids. J Biol Chem 17: 39953-39964. |
| [79] |
Li X, Lonard DM, O’Malley BW (2004) A contemporary understanding of progesterone receptor function. Mech Ageing Dev 125: 669-678. doi: 10.1016/j.mad.2004.04.007
|
| [80] | Mote PA, Graham JD, Clarke CL (2007) Progesterone receptor isoforms in normal and malignant breast. Ernst Schering Found Symp Proc 2007: 77-107. |
| [81] |
Zhu Y, Bond J, Thomas P (2003) Identification, classification, and partial characterization of genes in humans and other vertebrates homologous to a fish membrane progestin receptor. Proc Natl Acad Sci U S A 100: 2237-2242. doi: 10.1073/pnas.0436133100
|
| [82] | Kimura I, Nakayama Y, Konishi M, et al. (2012) Functions of MAPR (membrane-associated progesterone receptor) family members as heme/steroid-binding proteins. Curr Protein Pept Sci 13: 687-96 |
| [83] |
Su C, Cunningham RL, Rybalchenko N, et al. (2012) Progesterone increases the release of brain-derived neurotrophic factor from glia via progesterone receptor membrane component 1 (Pgrmc1)-dependent ERK5 signaling. Endocrinology 153: 4389-4400 doi: 10.1210/en.2011-2177
|
| [84] |
Xu J, Zeng C, Chu W, et al. (2011) Identification of the PGRMC1 protein complex as the putative sigma-2 receptor binding site. Nat Commun 2: 380. doi: 10.1038/ncomms1386
|
| [85] |
Kelder J, Azevedo R, Pang Y, et al. (2010) Comparison between steroid binding to membrane progesterone receptor alpha (mPR alpha) and to nuclear progesterone receptor: correlation with physicochemical properties assessed by comparative molecular field analysis and identification of mPR alpha specific agonists. Steroids 75: 314-322 doi: 10.1016/j.steroids.2010.01.010
|
| [86] | Zhu Y, Hanna RN, Schaaf MJ, et al. (2008) Candidates for membrane progestin receptors—past approaches and future challenges. Comp Biochem Physiol C Toxicol Pharmacol 14: 381-389. |
| [87] | Scarpin KM, Graham JD, Mote PA, et al. (2009) Progesterone action in human tissues: regulation by progesterone receptor (PR) isoform expression, nuclear positioning and coregulator expression. Nucl Recept Signal 7: e009 |
| [88] |
Thomas P (2008) Characteristics of membrane progestin receptor alpha (mPR alpha) and progesterone membrane receptor component 1 (PGMRC1) and their roles in mediating rapid progestin actions. Neuroendocrinol 29: 292-312. doi: 10.1016/j.yfrne.2008.01.001
|
| [89] |
Smith JL, Kupchak BR, Garitaonandia I, et al. (2008) Heterologous expression of human mPR alpha, mPR beta and mPR gamma in yeast confirms their ability to function as membrane progesterone receptors. Steroids 73: 1160-1173. doi: 10.1016/j.steroids.2008.05.003
|
| [90] |
Thomas P, Pang Y, Dong J, et al. (2007) Steroid and G protein binding characteristics of the sea trout and human progestin membrane receptor alpha subtypes and their evolutionary origins. Endocrinology 148: 705-718. doi: 10.1210/en.2006-0974
|
| [91] |
Zhu Y, Rice CD, Pang Y, et al. (2003) Cloning, expression, and characterization of a membrane progestin receptor and evidence it is an intermediary in meiotic maturation of fish oocytes. Proc Natl Acad Sci U S A 100: 2231-2236. doi: 10.1073/pnas.0336132100
|
| [92] |
Wang C, Liu Y, Cao JM (2014) Protein-Coupled Receptors: Extranuclear Mediators for the Non-Genomic Actions of Steroids. Int J Mol Sci 15: 15412-15425 doi: 10.3390/ijms150915412
|
| [93] |
Rey M, Coirini H (2015) Synthetic neurosteroids on brain protection. Neural Regen Res 10: 17-21 doi: 10.4103/1673-5374.150640
|
| [94] | Singh M, Su C, Ng S (2013) Non-genomic mechanisms of progesterone action in the brain. Front Neurosci 7: 159. |
| [95] |
Lonard DM, Lanz RB, O’Malley BW (2007) Nuclear receptor coregulators and human disease. Endocr Rev 28: 575-587 doi: 10.1210/er.2007-0012
|
| [96] |
Tubbs C, Thomas P (2008) Functional characteristics of membrane progestin receptor alpha (mPR alpha) subtypes: a review with new data showing mPRalpha expression in seatrout sperm and its association with sperm motility. Steroids 73: 935-941. doi: 10.1016/j.steroids.2007.12.022
|
| [97] |
Tung L, Abdel-Hafiz H, Shen T, et al. (2006) Progesterone receptors (PR)-B and -A regulate transcription by different mechanisms: AF-3 exerts regulatory control over coactivator binding to PR-B. Mol Endocrinol 20: 2656-2670 doi: 10.1210/me.2006-0105
|
| [98] |
Molenda-Figueira HA, Murphy SD, Shea KL, et al. (2008) Steroid receptor coactivator-1 from brain physically interacts differentially with steroid receptor subtypes. Endocrinology 149: 5272-5279 doi: 10.1210/en.2008-0048
|
| [99] |
Heneghan AF, Connaghan-Jones KD, Miura MT, et al. (2007) Coactivator assembly at the promoter: efficient recruitment of SRC2 is coupled to cooperative DNA binding by the progesterone receptor. Biochemistry 46: 11023-11032. doi: 10.1021/bi700850v
|
| [100] | Giangrande PH, McDonnell DP (1999) The A and B isoforms of the human progesterone receptor: two functionally different transcription factors encoded by a single gene. Recent Prog Horm Res 54: 291-313. |
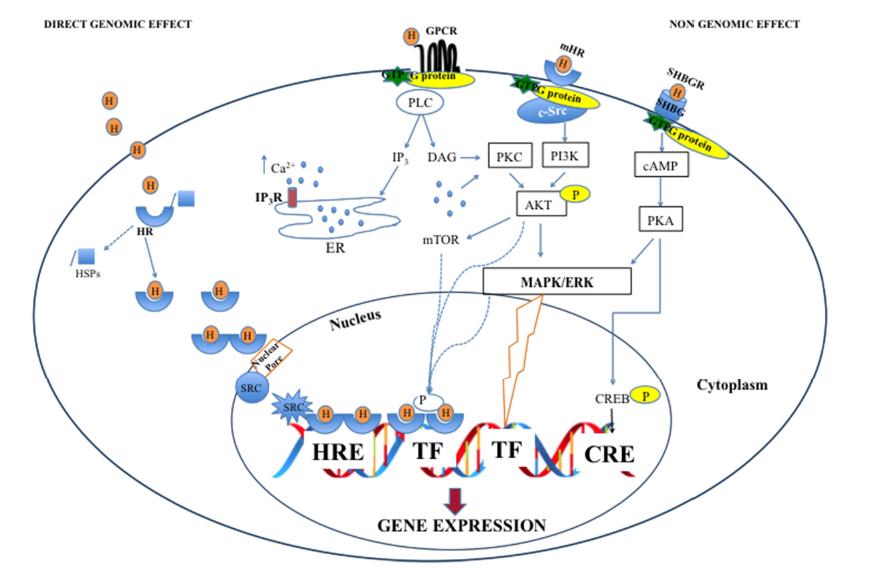
Figures(1)

Valentina Contrò, John R. Basile, Patrizia Proia. Sex steroid hormone receptors, their ligands, and nuclear and non-nuclear pathways[J]. AIMS Molecular Science, 2015, 2(3): 294-310. doi: 10.3934/molsci.2015.3.294







 DownLoad:
DownLoad: