Citation: Akshaj Pole, Manjari Dimri, Goberdhan P. Dimri. Oxidative stress, cellular senescence and ageing[J]. AIMS Molecular Science, 2016, 3(3): 300-324. doi: 10.3934/molsci.2016.3.300

| [1] | Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298-300. |
| [2] |
Harman D (2009) Origin and evolution of the free radical theory of aging: a brief personal history, 1954-2009. Biogerontology 10: 773-781. doi: 10.1007/s10522-009-9234-2

|
| [3] |
Perez VI, Bokov A, Van Remmen H, et al. (2009) Is the oxidative stress theory of aging dead? Biochim Biophys Acta 1790: 1005-1014. doi: 10.1016/j.bbagen.2009.06.003
|
| [4] | Jin K (2010) Modern Biological Theories of Aging. Aging Dis 1: 72-74. |
| [5] |
Jacob KD, Hooten NN, Trzeciak AR, et al. (2013) Markers of Oxidant Stress that are Clinically Relevant in Aging and Age-related Disease. Mech Ageing Dev 134: 139-157. doi: 10.1016/j.mad.2013.02.008
|
| [6] |
Gruber J, Fong S, Chen C-B, et al. (2013) Mitochondria-targeted antioxidants and metabolic modulators as pharmacological interventions to slow ageing. Biotechnol Adv 31: 563-592. doi: 10.1016/j.biotechadv.2012.09.005
|
| [7] | Vallyathan V, Shi X (1997) The role of oxygen free radicals in occupational and environmental lung diseases. Environ Health Perspect 105 Suppl 1: 165-177. |
| [8] |
Berneburg M, Gattermann N, Stege H, et al. (1997) Chronically ultraviolet-exposed human skin shows a higher mutation frequency of mitochondrial DNA as compared to unexposed skin and the hematopoietic system. Photochem Photobiol 66: 271-275. doi: 10.1111/j.1751-1097.1997.tb08654.x
|
| [9] |
Valko M, Rhodes CJ, Moncol J, et al. (2006) Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 160: 1-40. doi: 10.1016/j.cbi.2005.12.009
|
| [10] |
Ambrose JA, Barua RS (2004) The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol 43: 1731-1737. doi: 10.1016/j.jacc.2003.12.047
|
| [11] |
Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552: 335-344. doi: 10.1113/jphysiol.2003.049478
|
| [12] |
Schrader M, Fahimi HD (2006) Peroxisomes and oxidative stress. Biochim Biophys Acta 1763: 1755-1766. doi: 10.1016/j.bbamcr.2006.09.006
|
| [13] | Cheeseman KH, Slater TF (1993) An introduction to free radical biochemistry. Br Med Bull 49: 481-493. |
| [14] |
Krause KH (2007) Aging: a revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp Gerontol 42: 256-262. doi: 10.1016/j.exger.2006.10.011
|
| [15] |
Jiang F, Zhang Y, Dusting GJ (2011) NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev 63: 218-242. doi: 10.1124/pr.110.002980
|
| [16] |
Manea A (2010) NADPH oxidase-derived reactive oxygen species: involvement in vascular physiology and pathology. Cell Tissue Res 342: 325-339. doi: 10.1007/s00441-010-1060-y
|
| [17] |
Takac I, Schroder K, Brandes RP (2012) The Nox family of NADPH oxidases: friend or foe of the vascular system? Curr Hypertens Rep 14: 70-78. doi: 10.1007/s11906-011-0238-3
|
| [18] |
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245-313. doi: 10.1152/physrev.00044.2005
|
| [19] |
Rodriguez-Manas L, El-Assar M, Vallejo S, et al. (2009) Endothelial dysfunction in aged humans is related with oxidative stress and vascular inflammation. Aging Cell 8: 226-238. doi: 10.1111/j.1474-9726.2009.00466.x
|
| [20] |
Schroder K, Zhang M, Benkhoff S, et al. (2012) Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 110: 1217-1225. doi: 10.1161/CIRCRESAHA.112.267054
|
| [21] |
Chiu JJ, Chien S (2011) Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91: 327-387. doi: 10.1152/physrev.00047.2009
|
| [22] | Montezano AC, Touyz RM (2012) Oxidative stress, Noxs, and hypertension: experimental evidence and clinical controversies. Ann Med 44 Suppl 1: S2-16. |
| [23] |
Brand MD, Affourtit C, Esteves TC, et al. (2004) Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 37: 755-767. doi: 10.1016/j.freeradbiomed.2004.05.034
|
| [24] |
Muller FL, Liu Y, Van Remmen H (2004) Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279: 49064-49073. doi: 10.1074/jbc.M407715200
|
| [25] |
Madamanchi NR, Runge MS (2007) Mitochondrial dysfunction in atherosclerosis. Circ Res 100: 460-473. doi: 10.1161/01.RES.0000258450.44413.96
|
| [26] | Linnane AW, Marzuki S, Ozawa T, et al. (1989) Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1: 642-645. |
| [27] | Lee HC, Wei YH (2007) Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med (Maywood) 232: 592-606. |
| [28] |
Kwon MJ, Kim B, Lee YS, et al. (2012) Role of superoxide dismutase 3 in skin inflammation. J Dermatol Sci 67: 81-87. doi: 10.1016/j.jdermsci.2012.06.003
|
| [29] |
Kasapoglu M, Ozben T (2001) Alterations of antioxidant enzymes and oxidative stress markers in aging. Exp Gerontol 36: 209-220. doi: 10.1016/S0531-5565(00)00198-4
|
| [30] |
Marzani B, Felzani G, Bellomo RG, et al. (2005) Human muscle aging: ROS-mediated alterations in rectus abdominis and vastus lateralis muscles. Exp Gerontol 40: 959-965. doi: 10.1016/j.exger.2005.08.010
|
| [31] | Rodriguez-Capote K, Cespedes E, Arencibia R, et al. (1998) Indicators of oxidative stress in aging rat brain. The effect of nerve growth factor. Rev Neurol 27: 494-500. |
| [32] |
Lu CY, Lee HC, Fahn HJ, et al. (1999) Oxidative damage elicited by imbalance of free radical scavenging enzymes is associated with large-scale mtDNA deletions in aging human skin. Mutat Res 423: 11-21. doi: 10.1016/S0027-5107(98)00220-6
|
| [33] |
Treiber N, Maity P, Singh K, et al. (2011) Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue. Aging Cell 10: 239-254. doi: 10.1111/j.1474-9726.2010.00658.x
|
| [34] |
Mari M, Morales A, Colell A, et al. (2009) Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal 11: 2685-2700. doi: 10.1089/ars.2009.2695
|
| [35] |
Zhang H, Limphong P, Pieper J, et al. (2012) Glutathione-dependent reductive stress triggers mitochondrial oxidation and cytotoxicity. FASEB J 26: 1442-1451. doi: 10.1096/fj.11-199869
|
| [36] |
Gould NS, Min E, Gauthier S, et al. (2010) Aging adversely affects the cigarette smoke-induced glutathione adaptive response in the lung. Am J Respir Crit Care Med 182: 1114-1122. doi: 10.1164/rccm.201003-0442OC
|
| [37] | Doria E, Buonocore D, Focarelli A, et al. (2012) Relationship between human aging muscle and oxidative system pathway. Oxid Med Cell Longev 2012: 830257. |
| [38] |
Myung SK, Ju W, Cho B, et al. (2013) Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ 346: f10. doi: 10.1136/bmj.f10
|
| [39] |
Bjelakovic G, Nikolova D, Simonetti RG, et al. (2004) Antioxidant supplements for prevention of gastrointestinal cancers: a systematic review and meta-analysis. Lancet 364: 1219-1228. doi: 10.1016/S0140-6736(04)17138-9
|
| [40] |
Klotz LO, Sanchez-Ramos C, Prieto-Arroyo I, et al. (2015) Redox regulation of FoxO transcription factors. Redox Biol 6: 51-72. doi: 10.1016/j.redox.2015.06.019
|
| [41] |
Greer EL, Brunet A (2005) FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 24: 7410-7425. doi: 10.1038/sj.onc.1209086
|
| [42] |
Lin K, Dorman JB, Rodan A, et al. (1997) daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278: 1319-1322. doi: 10.1126/science.278.5341.1319
|
| [43] |
Ogg S, Paradis S, Gottlieb S, et al. (1997) The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389: 994-999. doi: 10.1038/40194
|
| [44] | Honda Y, Honda S (1999) The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J 13: 1385-1393. |
| [45] |
Kops GJ, Dansen TB, Polderman PE, et al. (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419: 316-321. doi: 10.1038/nature01036
|
| [46] |
van der Horst A, Burgering BM (2007) Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol 8: 440-450. doi: 10.1038/nrm2190
|
| [47] |
Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408: 239-247. doi: 10.1038/35041687
|
| [48] |
Honda Y, Honda S (2002) Oxidative stress and life span determination in the nematode Caenorhabditis elegans. Ann N Y Acad Sci 959: 466-474. doi: 10.1111/j.1749-6632.2002.tb02117.x
|
| [49] |
Ristow M, Schmeisser S (2011) Extending life span by increasing oxidative stress. Free Radic Biol Med 51: 327-336. doi: 10.1016/j.freeradbiomed.2011.05.010
|
| [50] |
Yoshioka T, Homma T, Meyrick B, et al. (1994) Oxidants induce transcriptional activation of manganese superoxide dismutase in glomerular cells. Kidney Int 46: 405-413. doi: 10.1038/ki.1994.288
|
| [51] |
Mesquita A, Weinberger M, Silva A, et al. (2010) Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc Natl Acad Sci U S A 107: 15123-15128. doi: 10.1073/pnas.1004432107
|
| [52] |
Yang W, Hekimi S (2010) A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol 8: e1000556. doi: 10.1371/journal.pbio.1000556
|
| [53] |
Schulz TJ, Zarse K, Voigt A, et al. (2007) Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 6: 280-293. doi: 10.1016/j.cmet.2007.08.011
|
| [54] |
Yun J, Finkel T (2014) Mitohormesis. Cell Metab 19: 757-766. doi: 10.1016/j.cmet.2014.01.011
|
| [55] | Yavari A, Javadi M, Mirmiran P, et al. (2015) Exercise-induced oxidative stress and dietary antioxidants. Asian J Sports Med 6: e24898. |
| [56] |
Elchuri S, Oberley TD, Qi W, et al. (2005) CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 24: 367-380. doi: 10.1038/sj.onc.1208207
|
| [57] |
Schriner SE, Linford NJ, Martin GM, et al. (2005) Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308: 1909-1911. doi: 10.1126/science.1106653
|
| [58] |
Lopez-Otin C, Blasco MA, Partridge L, et al. (2013) The hallmarks of aging. Cell 153: 1194-1217. doi: 10.1016/j.cell.2013.05.039
|
| [59] |
Esposito L, Raber J, Kekonius L, et al. (2006) Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci 26: 5167-5179. doi: 10.1523/JNEUROSCI.0482-06.2006
|
| [60] |
Torzewski M, Ochsenhirt V, Kleschyov AL, et al. (2007) Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 27: 850-857. doi: 10.1161/01.ATV.0000258809.47285.07
|
| [61] | Salmon AB, Flores LC, Li Y, et al. (2012) Reduction of glucose intolerance with high fat feeding is associated with anti-inflammatory effects of thioredoxin 1 overexpression in mice. Pathobiol Aging Age Relat Dis 2: 89-97. |
| [62] |
Shioji K, Kishimoto C, Nakamura H, et al. (2002) Overexpression of thioredoxin-1 in transgenic mice attenuates adriamycin-induced cardiotoxicity. Circulation 106: 1403-1409. doi: 10.1161/01.CIR.0000027817.55925.B4
|
| [63] |
Dizdaroglu M (2012) Oxidatively induced DNA damage: mechanisms, repair and disease. Cancer Lett 327: 26-47. doi: 10.1016/j.canlet.2012.01.016
|
| [64] |
Stadtman ER (2004) Role of oxidant species in aging. Curr Med Chem 11: 1105-1112. doi: 10.2174/0929867043365341
|
| [65] |
Yin H, Xu L, Porter NA (2011) Free radical lipid peroxidation: mechanisms and analysis. Chem Rev 111: 5944-5972. doi: 10.1021/cr200084z
|
| [66] |
de la Haba C, Palacio JR, Martinez P, et al. (2013) Effect of oxidative stress on plasma membrane fluidity of THP-1 induced macrophages. Biochim Biophys Acta 1828: 357-364. doi: 10.1016/j.bbamem.2012.08.013
|
| [67] |
Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25: 585-621. doi: 10.1016/0014-4827(61)90192-6
|
| [68] |
Dimri GP (2005) What has senescence got to do with cancer? Cancer Cell 7: 505-512. doi: 10.1016/j.ccr.2005.05.025
|
| [69] |
Tchkonia T, Zhu Y, van Deursen J, et al. (2013) Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 123: 966-972. doi: 10.1172/JCI64098
|
| [70] |
Banito A, Lowe SW (2013) A new development in senescence. Cell 155: 977-978. doi: 10.1016/j.cell.2013.10.050
|
| [71] |
Munoz-Espin D, Canamero M, Maraver A, et al. (2013) Programmed Cell Senescence during Mammalian Embryonic Development. Cell 155: 1104-1118. doi: 10.1016/j.cell.2013.10.019
|
| [72] |
Storer M, Mas A, Robert-Moreno A, et al. (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155: 1119-1130. doi: 10.1016/j.cell.2013.10.041
|
| [73] |
Bodnar AG, Ouellette M, Frolkis M, et al. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279: 349-352. doi: 10.1126/science.279.5349.349
|
| [74] |
Tan FC, Hutchison ER, Eitan E, et al. (2014) Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology 15: 643-660. doi: 10.1007/s10522-014-9532-1
|
| [75] |
Wang Z, Wei D, Xiao H (2013) Methods of cellular senescence induction using oxidative stress. Methods Mol Biol 1048: 135-144. doi: 10.1007/978-1-62703-556-9_11
|
| [76] |
Correia-Melo C, Hewitt G, Passos JF (2014) Telomeres, oxidative stress and inflammatory factors: partners in cellular senescence? Longev Healthspan 3: 1. doi: 10.1186/2046-2395-3-1
|
| [77] |
Hewitt G, Jurk D, Marques FD, et al. (2012) Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 3: 708. doi: 10.1038/ncomms1708
|
| [78] |
Itahana K, Campisi J, Dimri GP (2004) Mechanisms of cellular senescence in human and mouse cells. Biogerontology 5: 1-10. doi: 10.1023/B:BGEN.0000017682.96395.10
|
| [79] |
Dimri GP, Lee X, Basile G, et al. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92: 9363-9367. doi: 10.1073/pnas.92.20.9363
|
| [80] |
Narita M, Nunez S, Heard E, et al. (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703-716. doi: 10.1016/S0092-8674(03)00401-X
|
| [81] |
Alcorta DA, Xiong Y, Phelps D, et al. (1996) Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A 93: 13742-13747. doi: 10.1073/pnas.93.24.13742
|
| [82] |
Hara E, Smith R, Parry D, et al. (1996) Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol 16: 859-867. doi: 10.1128/MCB.16.3.859
|
| [83] | Coppe JP, Patil CK, Rodier F, et al. (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853-2868. |
| [84] |
Coppe JP, Desprez PY, Krtolica A, et al. (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99-118. doi: 10.1146/annurev-pathol-121808-102144
|
| [85] |
Coppe JP, Rodier F, Patil CK, et al. (2011) Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem 286: 36396-36403. doi: 10.1074/jbc.M111.257071
|
| [86] |
Chen Q, Fischer A, Reagan JD, et al. (1995) Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci U S A 92: 4337-4341. doi: 10.1073/pnas.92.10.4337
|
| [87] |
Parrinello S, Samper E, Krtolica A, et al. (2003) Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 5: 741-747. doi: 10.1038/ncb1024
|
| [88] |
Colavitti R, Finkel T (2005) Reactive oxygen species as mediators of cellular senescence. IUBMB Life 57: 277-281. doi: 10.1080/15216540500091890
|
| [89] |
d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, et al. (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194-198. doi: 10.1038/nature02118
|
| [90] |
Di Micco R, Fumagalli M, Cicalese A, et al. (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444: 638-642. doi: 10.1038/nature05327
|
| [91] | Passos JF, Nelson G, Wang C, et al. (2010) Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol 6: 347. |
| [92] |
Wiley CD, Velarde MC, Lecot P, et al. (2016) Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab 23: 303-314. doi: 10.1016/j.cmet.2015.11.011
|
| [93] |
Bracken AP, Kleine-Kohlbrecher D, Dietrich N, et al. (2007) The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev 21: 525-530. doi: 10.1101/gad.415507
|
| [94] |
Liu J, Cao L, Chen J, et al. (2009) Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 459: 387-392. doi: 10.1038/nature08040
|
| [95] |
Itahana K, Zou Y, Itahana Y, et al. (2003) Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol 23: 389-401. doi: 10.1128/MCB.23.1.389-401.2003
|
| [96] |
Acosta JC, Banito A, Wuestefeld T, et al. (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15: 978-990. doi: 10.1038/ncb2784
|
| [97] |
Kuilman T, Peeper DS (2009) Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 9: 81-94. doi: 10.1038/nrc2560
|
| [98] |
Passos JF, von Zglinicki T (2005) Mitochondria, telomeres and cell senescence. Exp Gerontol 40: 466-472. doi: 10.1016/j.exger.2005.04.006
|
| [99] |
Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120: 483-495. doi: 10.1016/j.cell.2005.02.001
|
| [100] |
Correia-Melo C, Marques FD, Anderson R, et al. (2016) Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35: 724-742. doi: 10.15252/embj.201592862
|
| [101] |
Ziegler DV, Wiley CD, Velarde MC (2015) Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell 14: 1-7. doi: 10.1111/acel.12287
|
| [102] |
Gomes AP, Price NL, Ling AJ, et al. (2013) Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155: 1624-1638. doi: 10.1016/j.cell.2013.11.037
|
| [103] |
Mouchiroud L, Houtkooper RH, Moullan N, et al. (2013) The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 154: 430-441. doi: 10.1016/j.cell.2013.06.016
|
| [104] |
Canto C, Gerhart-Hines Z, Feige JN, et al. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056-1060. doi: 10.1038/nature07813
|
| [105] |
Ruderman NB, Xu XJ, Nelson L, et al. (2010) AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab 298: E751-760. doi: 10.1152/ajpendo.00745.2009
|
| [106] |
Price NL, Gomes AP, Ling AJ, et al. (2012) SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 15: 675-690. doi: 10.1016/j.cmet.2012.04.003
|
| [107] | Zhang H, Ryu D, Wu Y, et al. (2016) NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science in press. |
| [108] |
Baker DJ, Wijshake T, Tchkonia T, et al. (2011) Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479: 232-236. doi: 10.1038/nature10600
|
| [109] |
Campisi J (2013) Aging, cellular senescence, and cancer. Annu Rev Physiol 75: 685-705. doi: 10.1146/annurev-physiol-030212-183653
|
| [110] |
Collado M, Blasco MA, Serrano M (2007) Cellular senescence in cancer and aging. Cell 130: 223-233. doi: 10.1016/j.cell.2007.07.003
|
| [111] |
Naylor RM, Baker DJ, van Deursen JM (2013) Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin Pharmacol Ther 93: 105-116. doi: 10.1038/clpt.2012.193
|
| [112] |
Childs BG, Durik M, Baker DJ, et al. (2015) Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 21: 1424-1435. doi: 10.1038/nm.4000
|
| [113] |
Passos JF, Saretzki G, Ahmed S, et al. (2007) Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol 5: e110. doi: 10.1371/journal.pbio.0050110
|
| [114] |
Blackburn EH, Epel ES, Lin J (2015) Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 350: 1193-1198. doi: 10.1126/science.aab3389
|
| [115] |
Fyhrquist F, Saijonmaa O, Strandberg T (2013) The roles of senescence and telomere shortening in cardiovascular disease. Nat Rev Cardiol 10: 274-283. doi: 10.1038/nrcardio.2013.30
|
| [116] |
Minamino T, Miyauchi H, Yoshida T, et al. (2004) The role of vascular cell senescence in atherosclerosis: antisenescence as a novel therapeutic strategy for vascular aging. Curr Vasc Pharmacol 2: 141-148. doi: 10.2174/1570161043476393
|
| [117] |
Navab M, Berliner JA, Watson AD, et al. (1996) The Yin and Yang of oxidation in the development of the fatty streak. A review based on the 1994 George Lyman Duff Memorial Lecture. Arterioscler Thromb Vasc Biol 16: 831-842. doi: 10.1161/01.ATV.16.7.831
|
| [118] |
Csiszar A, Wang M, Lakatta EG, et al. (2008) Inflammation and endothelial dysfunction during aging: role of NF-kappaB. J Appl Physiol (1985) 105: 1333-1341. doi: 10.1152/japplphysiol.90470.2008
|
| [119] |
Acosta JC, O'Loghlen A, Banito A, et al. (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006-1018. doi: 10.1016/j.cell.2008.03.038
|
| [120] |
Chien Y, Scuoppo C, Wang X, et al. (2011) Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev 25: 2125-2136. doi: 10.1101/gad.17276711
|
| [121] |
Freund A, Patil CK, Campisi J (2011) p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 30: 1536-1548. doi: 10.1038/emboj.2011.69
|
| [122] | Mowla SN, Perkins ND, Jat PS (2013) Friend or foe: emerging role of nuclear factor kappa-light-chain-enhancer of activated B cells in cell senescence. Onco Targets Ther 6: 1221-1229. |
| [123] |
Eren M, Boe AE, Murphy SB, et al. (2014) PAI-1-regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc Natl Acad Sci U S A 111: 7090-7095. doi: 10.1073/pnas.1321942111
|
| [124] | Kuro-o M (2008) Klotho as a regulator of oxidative stress and senescence. Biol Chem 389: 233-241. |
| [125] |
Sato S, Kawamata Y, Takahashi A, et al. (2015) Ablation of the p16(INK4a) tumour suppressor reverses ageing phenotypes of klotho mice. Nat Commun 6: 7035. doi: 10.1038/ncomms8035
|
| [126] |
Maekawa Y, Ishikawa K, Yasuda O, et al. (2009) Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappaB activation. Endocrine 35: 341-346. doi: 10.1007/s12020-009-9181-3
|
| [127] |
Luzi L, Confalonieri S, Di Fiore PP, et al. (2000) Evolution of Shc functions from nematode to human. Curr Opin Genet Dev 10: 668-674. doi: 10.1016/S0959-437X(00)00146-5
|
| [128] |
Migliaccio E, Giorgio M, Mele S, et al. (1999) The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 402: 309-313. doi: 10.1038/46311
|
| [129] |
Suski JM, Karkucinska-Wieckowska A, Lebiedzinska M, et al. (2011) p66Shc aging protein in control of fibroblasts cell fate. Int J Mol Sci 12: 5373-5389. doi: 10.3390/ijms12085373
|
| [130] |
Franzeck FC, Hof D, Spescha RD, et al. (2012) Expression of the aging gene p66Shc is increased in peripheral blood monocytes of patients with acute coronary syndrome but not with stable coronary artery disease. Atherosclerosis 220: 282-286. doi: 10.1016/j.atherosclerosis.2011.10.035
|
| [131] |
Cosentino F, Francia P, Camici GG, et al. (2008) Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein. Arterioscler Thromb Vasc Biol 28: 622-628. doi: 10.1161/ATVBAHA.107.156059
|
| [132] |
Carpi A, Menabo R, Kaludercic N, et al. (2009) The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta 1787: 774-780. doi: 10.1016/j.bbabio.2009.04.001
|
| [133] |
Morley JE, Armbrecht HJ, Farr SA, et al. (2012) The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer's disease. Biochim Biophys Acta 1822: 650-656. doi: 10.1016/j.bbadis.2011.11.015
|
| [134] | Nunomura A, Perry G, Aliev G, et al. (2001) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60: 759-767. |
| [135] |
Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10: 698-712. doi: 10.1038/nrd3505
|
| [136] | Casley CS, Canevari L, Land JM, et al. (2002) Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem 80: 91-100. |
| [137] | Swerdlow RH, Burns JM, Khan SM (2010) The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimers Dis 20 Suppl 2: S265-279. |
| [138] |
Bhat R, Crowe EP, Bitto A, et al. (2012) Astrocyte senescence as a component of Alzheimer's disease. PLoS One 7: e45069. doi: 10.1371/journal.pone.0045069
|
| [139] |
Streit WJ, Xue QS (2009) Life and death of microglia. J Neuroimmune Pharmacol 4: 371-379. doi: 10.1007/s11481-009-9163-5
|
| [140] |
Boccardi V, Pelini L, Ercolani S, et al. (2015) From cellular senescence to Alzheimer's disease: The role of telomere shortening. Ageing Res Rev 22: 1-8. doi: 10.1016/j.arr.2015.04.003
|
| [141] |
Dawson TM, Ko HS, Dawson VL (2010) Genetic animal models of Parkinson's disease. Neuron 66: 646-661. doi: 10.1016/j.neuron.2010.04.034
|
| [142] |
Ahlskog JE (2005) Challenging conventional wisdom: the etiologic role of dopamine oxidative stress in Parkinson's disease. Mov Disord 20: 271-282. doi: 10.1002/mds.20362
|
| [143] |
Ohtsuka C, Sasaki M, Konno K, et al. (2013) Changes in substantia nigra and locus coeruleus in patients with early-stage Parkinson's disease using neuromelanin-sensitive MR imaging. Neurosci Lett 541: 93-98. doi: 10.1016/j.neulet.2013.02.012
|
| [144] | Hattingen E, Magerkurth J, Pilatus U, et al. (2009) Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson's disease. Brain 132: 3285-3297. |
| [145] |
Srinivasan V, Cardinali DP, Srinivasan US, et al. (2011) Therapeutic potential of melatonin and its analogs in Parkinson's disease: focus on sleep and neuroprotection. Ther Adv Neurol Disord 4: 297-317. doi: 10.1177/1756285611406166
|
| [146] |
Corti O, Lesage S, Brice A (2011) What genetics tells us about the causes and mechanisms of Parkinson's disease. Physiol Rev 91: 1161-1218. doi: 10.1152/physrev.00022.2010
|
| [147] |
Perfeito R, Cunha-Oliveira T, Rego AC (2012) Revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease--resemblance to the effect of amphetamine drugs of abuse. Free Radic Biol Med 53: 1791-1806. doi: 10.1016/j.freeradbiomed.2012.08.569
|
| [148] |
Chinta SJ, Lieu CA, Demaria M, et al. (2013) Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson's disease? J Intern Med 273: 429-436. doi: 10.1111/joim.12029
|
| [149] |
Palmer AK, Tchkonia T, LeBrasseur NK, et al. (2015) Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 64: 2289-2298. doi: 10.2337/db14-1820
|
| [150] |
Ksiazek K, Passos JF, Olijslagers S, et al. (2008) Mitochondrial dysfunction is a possible cause of accelerated senescence of mesothelial cells exposed to high glucose. Biochem Biophys Res Commun 366: 793-799. doi: 10.1016/j.bbrc.2007.12.021
|
| [151] |
Tchkonia T, Morbeck DE, Von Zglinicki T, et al. (2010) Fat tissue, aging, and cellular senescence. Aging Cell 9: 667-684. doi: 10.1111/j.1474-9726.2010.00608.x
|
| [152] |
Moiseeva O, Deschenes-Simard X, St-Germain E, et al. (2013) Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 12: 489-498. doi: 10.1111/acel.12075
|
| [153] | Martin-Montalvo A, Mercken EM, Mitchell SJ, et al. (2013) Metformin improves healthspan and lifespan in mice. Nat Commun 4: 2192. |
| [154] | Glasauer A, Chandel NS (2014) Targeting antioxidants for cancer therapy. Biochem Pharmacol 92: 90-101. |
| [155] |
Reuter S, Gupta SC, Chaturvedi MM, et al. (2010) Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 49: 1603-1616. doi: 10.1016/j.freeradbiomed.2010.09.006
|
| [156] |
Trachootham D, Alexandre J, Huang P (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8: 579-591. doi: 10.1038/nrd2803
|
| [157] |
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646-674. doi: 10.1016/j.cell.2011.02.013
|
| [158] |
Collado M, Serrano M (2010) Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10: 51-57. doi: 10.1038/nrc2772
|
| [159] |
Campisi J (2011) Cellular senescence: putting the paradoxes in perspective. Curr Opin Genet Dev 21: 107-112. doi: 10.1016/j.gde.2010.10.005
|
| [160] |
Krtolica A, Parrinello S, Lockett S, et al. (2001) Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A 98: 12072-12077. doi: 10.1073/pnas.211053698
|
| [161] |
Davalos AR, Coppe JP, Campisi J, et al. (2010) Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev 29: 273-283. doi: 10.1007/s10555-010-9220-9
|
| [162] |
Sun Y, Campisi J, Higano C, et al. (2012) Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med 18: 1359-1368. doi: 10.1038/nm.2890
|
| [163] |
Achuthan S, Santhoshkumar TR, Prabhakar J, et al. (2011) Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J Biol Chem 286: 37813-37829. doi: 10.1074/jbc.M110.200675
|
| [164] |
Cahu J, Bustany S, Sola B (2012) Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis 3: e446. doi: 10.1038/cddis.2012.183
|
| [165] |
Hoare M, Narita M (2013) Transmitting senescence to the cell neighbourhood. Nat Cell Biol 15: 887-889. doi: 10.1038/ncb2811
|
| [166] |
Krishnamurthy J, Torrice C, Ramsey MR, et al. (2004) Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114: 1299-1307. doi: 10.1172/JCI22475
|
| [167] |
Sharpless NE, Sherr CJ (2015) Forging a signature of in vivo senescence. Nat Rev Cancer 15: 397-408. doi: 10.1038/nrc3960
|
| [168] |
Burd CE, Sorrentino JA, Clark KS, et al. (2013) Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 152: 340-351. doi: 10.1016/j.cell.2012.12.010
|
| [169] |
Demaria M, Ohtani N, Youssef SA, et al. (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31: 722-733. doi: 10.1016/j.devcel.2014.11.012
|
| [170] |
Cheng S, Rodier F (2015) Manipulating senescence in health and disease: emerging tools. Cell Cycle 14: 1613-1614. doi: 10.1080/15384101.2015.1039359
|
| [171] |
Baker DJ, Childs BG, Durik M, et al. (2016) Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530: 184-189. doi: 10.1038/nature16932
|
| [172] | Xu M, Palmer AK, Ding H, et al. (2015) Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 4: e12997. |
| [173] |
Xu M, Tchkonia T, Ding H, et al. (2015) JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A 112: E6301-6310. doi: 10.1073/pnas.1515386112
|
| [174] |
Zhu Y, Tchkonia T, Pirtskhalava T, et al. (2015) The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14: 644-658. doi: 10.1111/acel.12344
|
| [175] | Chang J, Wang Y, Shao L, et al. (2016) Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22: 78-83. |
| [176] |
Yosef R, Pilpel N, Tokarsky-Amiel R, et al. (2016) Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 7: 11190. doi: 10.1038/ncomms11190
|
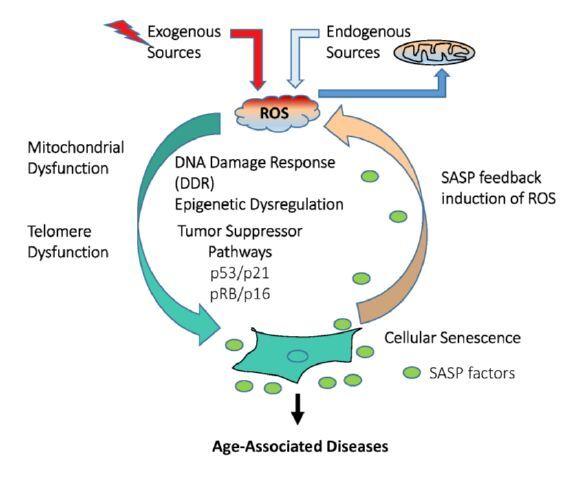
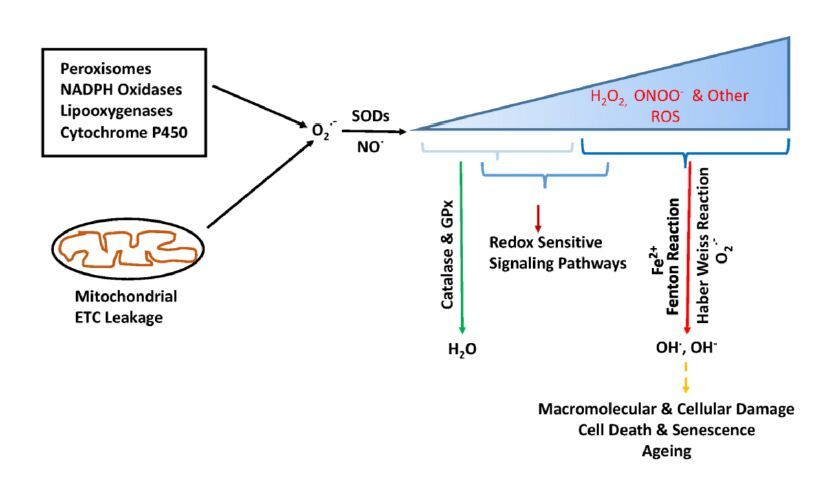
Figures(2)

Akshaj Pole, Manjari Dimri, Goberdhan P. Dimri. Oxidative stress, cellular senescence and ageing[J]. AIMS Molecular Science, 2016, 3(3): 300-324. doi: 10.3934/molsci.2016.3.300







 DownLoad:
DownLoad: