Citation: Naomi L. Pollock, Oscar Moran, Debora Baroni, Olga Zegarra-Moran, Robert C. Ford. Characterisation of the salmon cystic fibrosis transmembrane conductance regulator protein for structural studies[J]. AIMS Molecular Science, 2014, 1(4): 141-161. doi: 10.3934/molsci.2014.4.141

| [1] |
Plog S, Mundhenk L, Bothe MK, et al. (2010) Tissue and cellular expression patterns of porcine CFTR: similarities to and differences from human CFTR. J Histochem Cytochem 58: 785-797. doi: 10.1369/jhc.2010.955377

|
| [2] |
Crawford I, Maloney PC, Zeitlin PL, et al. (1991) Immunocytochemical localization of the cystic fibrosis gene product CFTR. P Natl Acad Sci USA 88: 9262-9266. doi: 10.1073/pnas.88.20.9262
|
| [3] |
Gadsby DC, Nairn AC (1999) Regulation of CFTR Cl- ion channels by phosphorylation and dephosphorylation. Adv Sec Messenger Phosphoprotein Res 33: 79-106. doi: 10.1016/S1040-7952(99)80006-8
|
| [4] | Gadsby DC, Nairn AC (1999) Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev 79: S77-S107. |
| [5] |
Kirk KL, Wang W (2011) A unified view of cystic fibrosis transmembrane conductance regulator (CFTR) gating: combining the allosterism of a ligand-gated channel with the enzymatic activity of an ATP-binding cassette (ABC) transporter. J Biol Chem 286: 12813-12819. doi: 10.1074/jbc.R111.219634
|
| [6] | Quinton PM, Reddy MM (2000) CFTR, a rectifying, non-rectifying anion channel? J Korean Med Sci 15 Suppl: S17-20. |
| [7] |
Goss CH, Ratjen F (2013) Update in cystic fibrosis 2012. Am J Resp Crit Care 187: 915-919. doi: 10.1164/rccm.201301-0184UP
|
| [8] |
Welsh MJ, Ramsey BW (1998) Research on cystic fibrosis: a journey from the Heart House. Am J Resp Crit Care 157: S148-154. doi: 10.1164/ajrccm.157.4.nhlbi-13
|
| [9] |
Hiroi J, McCormick SD (2012) New insights into gill ionocyte and ion transporter function in euryhaline and diadromous fish. Resp Physiol Neurobi 184: 257-268. doi: 10.1016/j.resp.2012.07.019
|
| [10] |
Christensen AK, Hiroi J, Schultz ET, et al. (2012) Branchial ionocyte organization and ion-transport protein expression in juvenile alewives acclimated to freshwater or seawater. J Exp Biol 215: 642-652. doi: 10.1242/jeb.063057
|
| [11] |
Chen JM, Cutler C, Jacques C, et al. (2001) A combined analysis of the cystic fibrosis transmembrane conductance regulator: implications for structure and disease models. Mol Biol Evol 18: 1771-1788. doi: 10.1093/oxfordjournals.molbev.a003965
|
| [12] |
Kiilerich P, Kristiansen K, Madsen SS (2007) Cortisol regulation of ion transporter mRNA in Atlantic salmon gill and the effect of salinity on the signaling pathway. J Endocrinol 194: 417-427. doi: 10.1677/JOE-07-0185
|
| [13] |
Nilsen TO, Ebbesson LO, Madsen SS, et al. (2007) Differential expression of gill Na+, K+-ATPase alpha- and beta-subunits, Na+, K+, 2Cl- cotransporter and CFTR anion channel in juvenile anadromous and landlocked Atlantic salmon Salmo salar. J Exp Biol 210: 2885-2896. doi: 10.1242/jeb.002873
|
| [14] |
Mio K, Ogura T, Mio M, et al. (2008) Three-dimensional reconstruction of human cystic fibrosis transmembrane conductance regulator chloride channel revealed an ellipsoidal structure with orifices beneath the putative transmembrane domain. J Biol Chem 283: 30300-30310. doi: 10.1074/jbc.M803185200
|
| [15] |
Rosenberg MF, O'Ryan LP, Hughes G, et al. (2011) The cystic fibrosis transmembrane conductance regulator (CFTR): three-dimensional structure and localization of a channel gate. J Biol Chem 286: 42647-42654. doi: 10.1074/jbc.M111.292268
|
| [16] |
Zhang L, Aleksandrov LA, Riordan JR, et al. (2011) Domain location within the cystic fibrosis transmembrane conductance regulator protein investigated by electron microscopy and gold labelling. BBA-Biomembranes 1808: 399-404. doi: 10.1016/j.bbamem.2010.08.012
|
| [17] |
Awayn NH, Rosenberg MF, Kamis AB, et al. (2005) Crystallographic and single-particle analyses of native- and nucleotide-bound forms of the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Biochem Soc T 33: 996-999. doi: 10.1042/BST20050996
|
| [18] |
Lewis HA, Buchanan SG, Burley SK, et al. (2004) Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J 23: 282-293. doi: 10.1038/sj.emboj.7600040
|
| [19] |
Thibodeau PH, Brautigam CA, Machius M, et al. (2005) Side chain and backbone contributions of Phe508 to CFTR folding. Nat Struct Mol Biol 12: 10-16. doi: 10.1038/nsmb881
|
| [20] |
Galeno L, Galfre E, Moran O (2011) Small-angle X-ray scattering study of the ATP modulation of the structural features of the nucleotide binding domains of the CFTR in solution. Eur Biophys J 40: 811-824. doi: 10.1007/s00249-011-0692-5
|
| [21] |
Galfre E, Galeno L, Moran O (2012) A potentiator induces conformational changes on the recombinant CFTR nucleotide binding domains in solution. Cell Mol Life Sci 69: 3701-3713. doi: 10.1007/s00018-012-1049-7
|
| [22] |
Marasini C, Galeno L, Moran O (2013) A SAXS-based ensemble model of the native and phosphorylated regulatory domain of the CFTR. Cell Mol Life Sci 70: 923-933. doi: 10.1007/s00018-012-1172-5
|
| [23] |
Hudson RP, Chong PA, Protasevich, II, et al. (2012) Conformational changes relevant to channel activity and folding within the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 287: 28480-28494. doi: 10.1074/jbc.M112.371138
|
| [24] |
Huang P, Liu Q, Scarborough GA (1998) Lysophosphatidylglycerol: a novel effective detergent for solubilizing and purifying the cystic fibrosis transmembrane conductance regulator. Anal biochem 259: 89-97. doi: 10.1006/abio.1998.2633
|
| [25] |
Wiener MC (2004) A pedestrian guide to membrane protein crystallization. Methods 34: 364-372. doi: 10.1016/j.ymeth.2004.03.025
|
| [26] |
Carpenter EP, Beis K, Cameron AD, et al. (2008) Overcoming the challenges of membrane protein crystallography. Curr Opin Struc Biol 18: 581-586. doi: 10.1016/j.sbi.2008.07.001
|
| [27] |
Dobrovetsky E, Menendez J, Edwards AM, et al. (2007) A robust purification strategy to accelerate membrane proteomics. Methods 41: 381-387. doi: 10.1016/j.ymeth.2006.08.009
|
| [28] |
Granseth E, Seppala S, Rapp M, et al. (2007) Membrane protein structural biology--how far can the bugs take us? Mol Membr Biol 24: 329-332. doi: 10.1080/09687680701413882
|
| [29] |
Lewinson O, Lee AT, Rees DC (2008) The funnel approach to the precrystallization production of membrane proteins. J Mol Biol 377: 62-73. doi: 10.1016/j.jmb.2007.12.059
|
| [30] |
Graeslund S (2008) Protein production and purification. Nat Meth 5: 135-146. doi: 10.1038/nmeth.f.202
|
| [31] |
Mancia F, Love J (2010) High-throughput expression and purification of membrane proteins. J Struct Biol 172: 85-93. doi: 10.1016/j.jsb.2010.03.021
|
| [32] |
Aller SG, Yu J, Ward A, et al. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323: 1718-1722. doi: 10.1126/science.1168750
|
| [33] |
Kawate T, Gouaux E (2006) Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14: 673-681. doi: 10.1016/j.str.2006.01.013
|
| [34] |
Sonoda Y, Cameron A, Newstead S, et al. (2010) Tricks of the trade used to accelerate high-resolution structure determination of membrane proteins. FEBS Lett 584: 2539-2547. doi: 10.1016/j.febslet.2010.04.015
|
| [35] |
Sonoda Y, Newstead S, Hu NJ, et al. (2011) Benchmarking membrane protein detergent stability for improving throughput of high-resolution X-ray structures. Structure 19: 17-25. doi: 10.1016/j.str.2010.12.001
|
| [36] |
Drew D, Newstead S, Sonoda Y, et al. (2008) GFP-based optimization scheme for the overexpression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae. Nat Protoc 3: 784-798. doi: 10.1038/nprot.2008.44
|
| [37] |
Newstead S, Kim H, von Heijne G, et al. (2007) High-throughput fluorescent-based optimization of eukaryotic membrane protein overexpression and purification in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 104: 13936-13941. doi: 10.1073/pnas.0704546104
|
| [38] | Clark KM, Fedoriw N, Robinson K, et al. Purification of transmembrane proteins from Saccharomyces cerevisiae for X-ray crystallography. Protein Expres Purif 71: 207-223. |
| [39] |
Slotboom DJ, Duurkens RH, Olieman K, et al. (2008) Static light scattering to characterize membrane proteins in detergent solution. Methods 46: 73-82. doi: 10.1016/j.ymeth.2008.06.012
|
| [40] | Ouano AC, Kaye W (1974) Gel-permeation chromatography: X. Molecular weight detection by low-angle laser light scattering. J Polym Sci: Polym Chem Edit 12: 1151-1162. |
| [41] |
Miller JL, Tate CG (2011) Engineering an ultra-thermostable beta(1)-adrenoceptor. J Mol Biol 413: 628-638. doi: 10.1016/j.jmb.2011.08.057
|
| [42] |
Shibata Y, White JF, Serrano-Vega MJ, et al. (2009) Thermostabilization of the neurotensin receptor NTS1. J Mol Biol 390: 262-277. doi: 10.1016/j.jmb.2009.04.068
|
| [43] |
Tate CG, Schertler GF (2009) Engineering G protein-coupled receptors to facilitate their structure determination. Curr Opin Struc Biol 19: 386-395. doi: 10.1016/j.sbi.2009.07.004
|
| [44] |
Warne T, Serrano-Vega MJ, Tate CG, et al. (2009) Development and crystallization of a minimal thermostabilised G protein-coupled receptor. Protein Expres Purif 65: 204-213. doi: 10.1016/j.pep.2009.01.014
|
| [45] |
Aleksandrov AA, Kota P, Cui L, et al. (2012) Allosteric modulation balances thermodynamic stability and restores function of DeltaF508 CFTR. J Mol Biol 419: 41-60. doi: 10.1016/j.jmb.2012.03.001
|
| [46] |
Huang P, Stroffekova K, Cuppoletti J, et al. (1996) Functional expression of the cystic fibrosis transmembrane conductance regulator in yeast. Biochim Biophys Acta 1281: 80-90. doi: 10.1016/0005-2736(96)00032-6
|
| [47] |
Bear CE, Li CH, Kartner N, et al. (1992) Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 68: 809-818. doi: 10.1016/0092-8674(92)90155-6
|
| [48] | Kogan I, Ramjeesingh M, Li C, et al. (2002) Studies of the molecular basis for cystic fibrosis using purified reconstituted CFTR protein. Method Mol Med 70: 143-157. |
| [49] |
Bai J, Swartz DJ, Protasevich, II, et al. (2011) A gene optimization strategy that enhances production of fully functional P-glycoprotein in Pichia pastoris. PloS One 6: e22577. doi: 10.1371/journal.pone.0022577
|
| [50] | O'Ryan L, Rimington T, Cant N, et al. (2012) Expression and purification of the cystic fibrosis transmembrane conductance regulator protein in Saccharomyces cerevisiae. J Vis Exp e3860. |
| [51] | Pollock N, Cant N, Rimington T, et al. (2014) Purification of the cystic fibrosis transmembrane conductance regulator protein expressed in Saccharomyces cerevisiae. J Vis Exp e51447. |
| [52] |
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Method 9: 671-675. doi: 10.1038/nmeth.2089
|
| [53] | Sievers F, Wilm A, Dineen D, et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539. |
| [54] |
Sievers F, Higgins DG (2014) Clustal Omega, accurate alignment of very large numbers of sequences. Methods in molecular biology 1079: 105-116. doi: 10.1007/978-1-62703-646-7_6
|
| [55] |
Dawson RJP, Locher KP (2007) Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett 581: 935-938. doi: 10.1016/j.febslet.2007.01.073
|
| [56] | Hammersley A, Svensson S, Hanfland M, et al. (1996) Two-Dimensional Detector Software: From Real Detector to Idealised Image or Two-Theta Scan. High Pressure Res 14: 325-348. |
| [57] | Mateu L, Luzzati V, Vargas R, et al. (1990) Order-disorder phenomena in myelinated nerve sheaths. II. The structure of myelin in native and swollen rat sciatic nerves and in the course of myelinogenesis. J Mol Biol 215: 385-402. |
| [58] |
Luzzati V, Tardieu A (1980) Recent developments in solution x-ray scattering. Annu Rev Biophys Bioeng 9: 1-29. doi: 10.1146/annurev.bb.09.060180.000245
|
| [59] |
Petoukhov M, Svergun D (2007) Analysis of X-ray and neutron scattering from biomacromolecular solutions. Curr Opin Struc Biol 17: 562-571. doi: 10.1016/j.sbi.2007.06.009
|
| [60] | Guinier A, Fournet G (1955) Small angle scattering of x-rays. New York: Wiley. |
| [61] | Feigin L, Svergun D (1987) Structure analysis by small-angle x.ray and neutron scattering. New York, London: Plenum Press. |
| [62] |
Svergun D (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 25: 495-503. doi: 10.1107/S0021889892001663
|
| [63] |
Dawson RJ, Locher KP (2006) Structure of a bacterial multidrug ABC transporter. Nature 443: 180-185. doi: 10.1038/nature05155
|
| [64] |
Franke D, Svergun D (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr 42: 342-346. doi: 10.1107/S0021889809000338
|
| [65] |
Tian C, Vanoye CG, Kang C, et al. (2007) Preparation, functional characterization, and NMR studies of human KCNE1, a voltage-gated potassium channel accessory subunit associated with deafness and long QT syndrome. Biochemistry 46: 11459-11472. doi: 10.1021/bi700705j
|
| [66] |
Oliver RC, Lipfert J, Fox DA, et al. (2013) Dependence of micelle size and shape on detergent alkyl chain length and head group. PloS One 8: e62488. doi: 10.1371/journal.pone.0062488
|
| [67] |
Yang Z, Wang C, Zhou Q, et al. (2014) Membrane protein stability can be compromised by detergent interactions with the extramembranous soluble domains. Protein Sci 23: 769-789. doi: 10.1002/pro.2460
|
| [68] |
Gulati S, Jamshad M, Knowles TJ, et al. (2014) Detergent-free purification of ABC (ATP-binding-cassette) transporters. Biochem J 461: 269-278. doi: 10.1042/BJ20131477
|
| [69] |
Lyman CP (1968) Body temperature of exhausted salmon. Copeia 1968: 631-633. doi: 10.2307/1442045
|
| [70] | Behrisch HW (1969) Temperature and the regulation of enzyme activity in poikilotherms. Fructose diphosphatase from migrating salmon. Biochem J 115: 687-696. |
| [71] | Handeland SO, Berge Ö, Björnsson BT, et al. (2000) Seawater adaptation by out-of-season Atlantic salmon (Salmo salar L.) smolts at different temperatures. Aquaculture 181: 377-396. |
| [72] |
Hsu HH, Lin LY, Tseng YC, et al. (2014) A new model for fish ion regulation: identification of ionocytes in freshwater- and seawater-acclimated medaka (Oryzias latipes). Cell Tissue Res 357: 225-243. doi: 10.1007/s00441-014-1883-z
|
| [73] | Moorman BP, Inokuchi M, Yamaguchi Y, et al. (2014) The osmoregulatory effects of rearing Mozambique tilapia in a tidally changing salinity. Gen Comp Endocrinol [in press]. |
| [74] |
Sucre E, Bossus M, Bodinier C, et al. (2013) Osmoregulatory response to low salinities in the European sea bass embryos: a multi-site approach. J Comp Physiol B 183: 83-97. doi: 10.1007/s00360-012-0687-2
|
| [75] |
Guggino WB, Stanton BA (2006) New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol 7: 426-436. doi: 10.1038/nrm1949
|
| [76] |
Venerando A, Franchin C, Cant N, et al. (2013) Detection of phospho-sites generated by protein kinase CK2 in CFTR: mechanistic aspects of Thr1471 phosphorylation. PloS One 8: e74232. doi: 10.1371/journal.pone.0074232
|
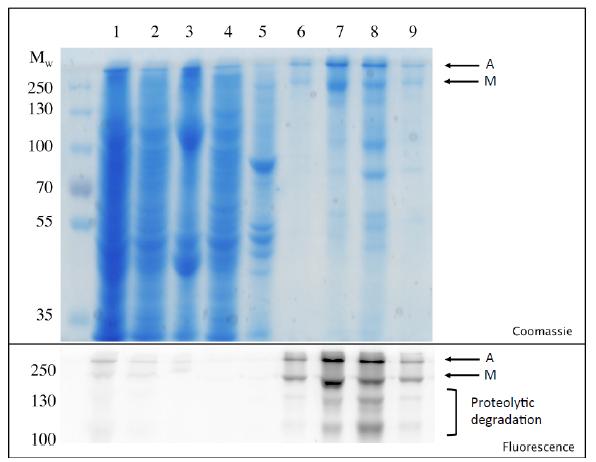
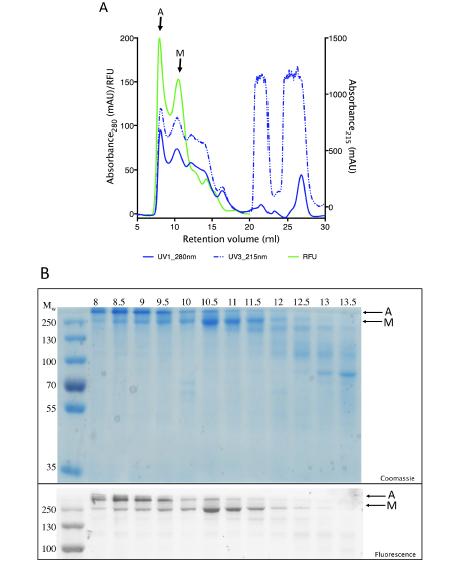
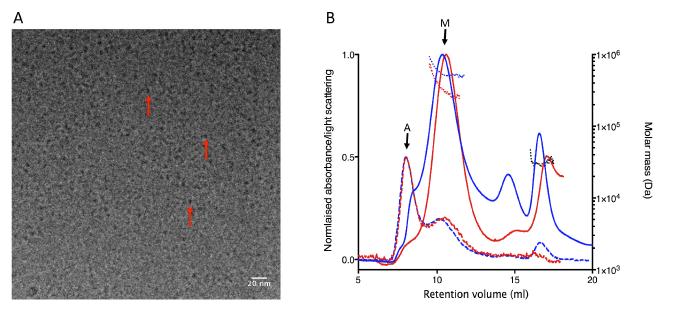

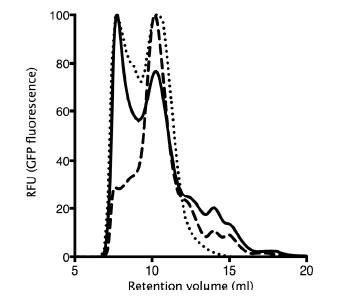
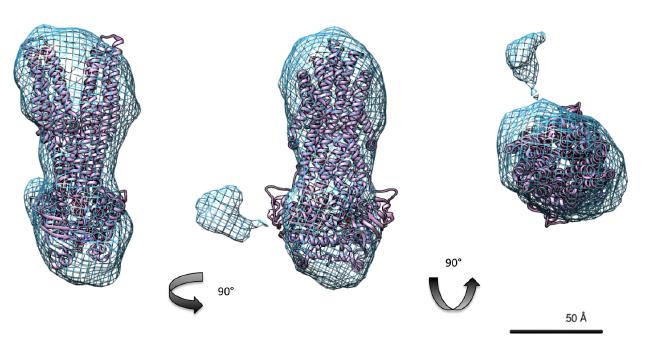
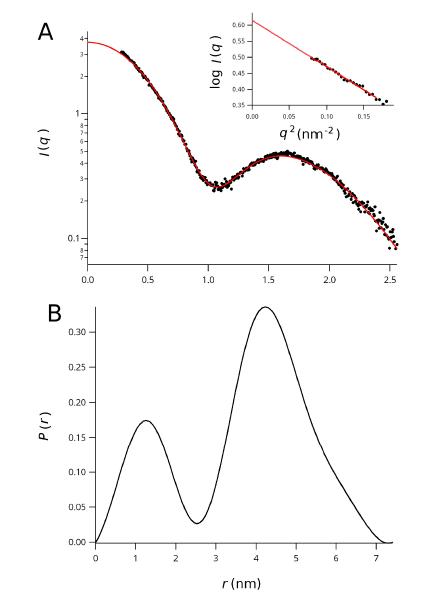
Figures(7) / Tables(2)

Naomi L. Pollock, Oscar Moran, Debora Baroni, Olga Zegarra-Moran, Robert C. Ford. Characterisation of the salmon cystic fibrosis transmembrane conductance regulator protein for structural studies[J]. AIMS Molecular Science, 2014, 1(4): 141-161. doi: 10.3934/molsci.2014.4.141







 DownLoad:
DownLoad: