Citation: Jiajun Zhang, Tianshou Zhou. Stationary moments, distribution conjugation and phenotypic regions in stochastic gene transcription[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 6134-6166. doi: 10.3934/mbe.2019307

| [1] | L. Cai, N. Friedman and X. S. Xie, Stochastic protein expression in individual cells at the single molecule level, Nature, 440 (2006), 358–362. |
| [2] | J. R. Chubb and T. B. Liverpool, Bursts and pulses: insights from single cell studies into transcriptional mechanisms, Curr. Opin. Genet. Dev., 20 (2010), 478–84. |
| [3] | D. R. Larson, What do expression dynamics tell us about the mechanism of transcription? Curr. Opin. Genet. Dev., 21 (2011), 591–599. |
| [4] | A. Sanchez and I. Golding, Genetic determinants and cellular constraints in noisy gene expression, Science, 342 (2013), 1188–1193. |
| [5] | J. Yu, J. Xiao, X. J. Ren, et al., Probing gene expression in live cells, one protein molecule at a time, Science, 311 (2006), 1600–1603. |
| [6] | W. J. Blake , M. Karn, C. R. Cantor, et al., Noise in eukaryotic gene expression, Nature, 422 (2003), 633–637. |
| [7] | J. R. Chubb, T. Trcek, S. M. Shenoy, et al., Transcriptional pulsing of a developmental gene, Curr. Biol., 16 (2006), 1018–1025. |
| [8] | I. Golding, J. Paulsson, S. M. Zawilski, et al., Real-time kinetics of gene activity in individual bacteria, Cell, 123 (2005), 1025–1036. |
| [9] | A. Raj, C. S. Peskin, D. Tranchina, et al., Stochastic mRNA synthesis in mammalian cells, PLoS. Biol., 4 (2006), e309. |
| [10] | H. Boeger, J. Griesenbeck and R. D. Kornberg, Nucleosome retention and the stochastic nature of promoter chromatin remodeling for transcription, Cell, 133 (2008), 716–726. |
| [11] | C. V. Harper, B. Finkenstadt, D. J. Woodcock, et al., Dynamic analysis of stochastic transcription cycles, PLoS Biol., 9 (2011), e1000607. |
| [12] | C. Mao, C. R. Brown, S. F. Dong, et al., Quantitative analysis of the transcription control mechanism, Mol. Syst. Biol., 6 (2010), 431. |
| [13] | L. Mariani, E. G. Schulz, E. Falkovskaia, et al., Short-term memory in gene induction reveals the regulatory principle behind stochastic IL-4 expression, Mol. Syst. Biol., 6 (2010), 359. |
| [14] | K. Miller-Jensen, S. S. Dey, D. V. Schaffer, et al., Varying virulence: epigenetic control of expression noise and disease processes, Trends Biotechnol, 29 (2011), 517–525. |
| [15] | J. Raser and E. O'Shea, Control of stochasticity in eukaryotic gene expression, Science, 304 (2003), 1811–1814. |
| [16] | D. M. Suter, N. Molina, D. Gatfield, et al., Mammalian genes are transcribed with widely different bursting kinetics, Science, 332 (2011), 472–474. |
| [17] | G. Hager, C. Elbi, T. A. Johnson, et al., Chromatin dynamics and the evolution of alternate promoter states, Chromosome Res., 14 (2006), 107–116. |
| [18] | T. J. Stasevich and J. G. McNally, Assembly of the transcription machinery: ordered and stable, random and dynamic, or both? Chromosoma, 120 (2011), 533–545. |
| [19] | L. Weinberger, Y. Voichek, I. Tirosh, et al., Expression noise and acetylation profiles distinguish HDAC functions, Mol. Cell, 47 (2012), 193–202. |
| [20] | A. Schwabe, K. N. Rybakova and F. J. Bruggeman, Transcription stochasticity of complex gene regulation models, Biophys. J., 103 (2012), 1152–1161. |
| [21] | W. J. Blake, G. Balázsi, M. A. Kohanski, et al., Phenotypic consequences of promoter-mediated transcriptional noise, Mol. Cell, 6 (2006), 853–865. |
| [22] | M. Dobrzynski and F. J. Bruggeman, Elongation dynamics shape bursty transcription and translation, Proc. Natl. Acad. Sci. USA, 106 (2009), 2583–2588. |
| [23] | A. Sanchez, H. G. Garcia, D. Jones, et al., Effect of promoter architecture on the cell-to-cell variability in gene expression, PLoS. Comput. Biol., 7 (2011), e1001100. |
| [24] | J. J. Zhang, L. N. Chen and T. S. Zhou, Analytical distribution and tunability of noise in a model of promoter progress, Biophys. J., 102 (2012), 1247–1257. |
| [25] | J. J. Zhang and T. S. Zhou, Promoter-mediated transcriptional dynamics, Biophys. J., 106 (2014), 479–488. |
| [26] | J. M. G. Vilar and L. Saiz, CplexA: a mathematica package to study macromolecular-assembly control of gene expression, Bioinform, 26 (2010), 2060–2061. |
| [27] | G. Hornung, R. Bar-Ziv, D. Rosin, et al., Noise-mean relationship in mutated promoters, Genome Res., 22 (2012), 2409–2417. |
| [28] | A..Halme, S. Bumgarner, C. Styles, et al., Genetic and epigenetic regulation of the FLO gene family generates cell-surface variation in yeast, Cell, 116 (2004), 405–415. |
| [29] | L. M. Octavio, K. Gedeon and N. Maheshri, Epigenetic and conventional regulation is distributed among activators of FLO11 allowing tuning of population-level heterogeneity in its expression, PLoS. Genet., 5 (2009), e1000673. |
| [30] | L. Bintu, N. E. Buchler, H. G. Garcia, et al., Transcriptional regulation by the numbers: models, Curr. Opin. Genet. Dev., 15 (2005), 116–124. |
| [31] | D. Huh and J. Paulsson, Non-genetic heterogeneity from stochastic partitioning at cell division., Nat. Genet., 43 (2011), 95–100. |
| [32] | T. L. To and N. Maheshri, Noise can induce bimodality in positive transcriptional feedback loops without bistability, Science, 327 (2010), 1142–1145. |
| [33] | R. Skupsky, J. C. Burnett, J. E. Foley, et al., HIV promoter integration site primarily modulates transcriptional burst size rather than frequency, PLoS. Comput. Biol., 6 (2010), e1000952. |
| [34] | L. S. Weinberger, J. C. Burnett, J. E. Toettcher, et al., Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity, Cell, 122 (2005), 169–182. |
| [35] | Q. L. Wang and T. S. Zhou, Alternative-splicing-mediated gene expression, Phys. Rev. E., 89 (2014), 012713. |
| [36] | A. Ochab-Marcinek and M. Tabaka, Bimodal gene expression in noncooperative regulatory systems, Proc. Natl. Acad. Sci. USA, 107 (2010), 22096–22101. |
| [37] | N. Friedman, L. Cai and X. S. Xie, Linking stochastic dynamics to population- distribution: an analytical framework of gene expression, Phys. Rev. Lett., 97 (2006), 168302. |
| [38] | P. J. Liu, Z. J. Yuan, L. F. Huang, et al., Roles of factorial noise in inducing bimodal gene expression, Phys. Rev. E., 91 (2015), 062706. |
| [39] | V. Shahrezaei and P. S. Swain, Analytical distributions for stochastic gene expression, Proc. Natl. Acad. Sci. USA, 105 (2008), 17256–17261. |
| [40] | B. Bodenmiller, E. R. Zunder, R. Finck, et al., Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators, Nat. Biotechnol, 30 (2012), 858. |
| [41] | N. Rimon and M. Schuldiner, Getting the whole picture: combining throughput with content in microscopy, J. Cell. Sci., 124 (2011), 3743. |
| [42] | Y. Liu, A Beyer and R. Aebersold, On the dependency of cellular protein levels on mRNA abundance, Cell, 165 (2016), 535–550. |
| [43] | J. J. Zhang, L. F. Huang and T. S. Zhou, A moment-convergence method for stochastic analysis of biochemical reaction networks, J. Chem. Phys., 144 (2016), 018620. |
| [44] | L. J. Slater, Confluent Hypergeometric Functions, Cambridge University Press, Cambridge, 1960. |
| [45] | Y. He and E. Barkai, Super- and sub-Poissonian photon statistics for single molecule spectroscopy, J. Chem. Phys., 122 (2005), 184703. |
| [46] | J. M. Pedraza and J. Paulsson, Effects of molecular memory and bursting on fluctuations in gene expression, Science, 319 (2008), 339–343. |
| [47] | L. F. Huang, Z. J. Yuan, P. J. Liu, et al., Feedback-induced counterintuitive correlations of gene expression noise with bursting kinetics, Phys. Rev. E., 90 (2014), 052702. |
| [48] | J. Paulsson, Models of stochastic gene expression, Phys. Life. Rev., 2 (2005), 157–175. |
| [49] | J. Peccoud and B. Ycart, Markovian modelling of gene product synthesis, Theor. Popul. Biol., 48 (1995), 222–234. |
| [50] | J. Tao and R. V. Kulkarni, Intrinsic noise in stochastic models of gene expression with molecular memory and bursting, Phys. Rev. Lett., 106 (2011), 058102. |
| [51] | T. B. Kepler and T. C. Elston, Stochasticity in transcriptional regulation: origins, consequences, and mathematical representations, Biophys. J., 81 (2001), 3116–3036. |
| [52] | B. J. Daigle, Jr. M. Soltani, L. R. Petzold, et al., Inferring single-cell gene expression mechanisms using stochastic simulation, Bioinformatics, 31 (2015), 1428–1435. |
| [53] | B. Zoller, D. Nicolas, N. Molina, et al., Structure of silent transcription intervals and noise characteristics of mammalian genes, Mol. Syst. Biol., 11 (2015), 823. |
| [54] | L Liu, B. R. K. Kashyap and J. G. C. Templeton, On the GIX/G/∞ system, J. Appl. Probab., 27 (1990), 671–683. |
| [55] | M. Thattai, Universal Poisson statistics of mRNAs with complex decay pathways, Biophys. J., 110 (2016), 301–305. |
| [56] | A. R. Stinchcombe, C. S. Peskin and D. Tranchina, Population density approach for discrete mRNA distributions in generalized switching models for stochastic gene expression, Phys. Rev. E., 85 (2012), 061919. |
| [57] | T. S. Zhou and J. J. Zhang, Analytical results for a multistate gene model, SIAM. J. Appl. Math., 72 (2012), 789–818. |
| [58] | A. Sánchez, S. Choubey and J. Kondev, Stochastic models of transcription: From single molecules to single cells, Methods, 62 (2013), 13–25. |
| [59] | J. Garcia-Bernardo and M. J. Dunlop, Phenotypic diversity using bimodal and unimodal expression of stress response proteins, Biophys. J., 110 (2016), 2278–2287. |
| [60] | D. T. Gillespie, A general method for numerically simulating the stochastic time evolution of coupled chemial reactions, J. Comput. Phys., 22 (1976), 403. |
| [61] | Q. L. Wang and T. S. Zhou, Dynamical analysis of mCAT2 gene models with CTN-RNA nuclear retention, Phys. Biol., 12 (2015), 016010. |
| [62] | H. H. Wang, Z. J. Yuan, P. J. Liu, et al., Division time-based amplifiers for gene expression, Mol. Biosyst., 11 (2015), 2417–2428. |
| [63] | M. Sturrock, P. J. Murray, A. Matzavinos, et al., Mean field analysis of a spatial stochastic model of a gene regulatory network, J. Math. Biol., 71 (2015), 921–959. |
| [64] | M. Sturrock, S. Li and V. Shahrezaei, The influence of nuclear compartmentalisation on stochastic dynamics of self-repressing gene expression, J. Theor. Biol., 424 (2017), 55–72. |
| [65] | N. Balakrishnan, N. L. Johnson and S. Kotz, A note on relationships between moments, central moments and cumulants from multivariate distributions, Stat. Prob. Lett. 39 (1998), 49–54. |
| [66] | N. Kumar, A. Singh and R. V. Kulkarni, Transcriptional Bursting in Gene Expression: Analytical Results for General Stochastic Models, PLoS. Comput. Biol., 11 (2015), e1004292. |
| [67] | L. Takács, An Introduction to queueing theory, Oxford University Press, New York, 1962. |
| [68] | H. Xu, S. O. Skinner, A. M. Sokac, et al., Stochastic kinetics of nascent RNA, Phys. Rev. Lett., 117 (2016), 128101. |
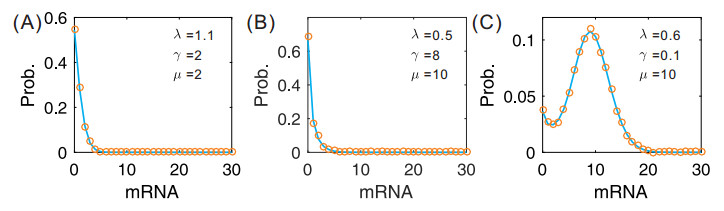
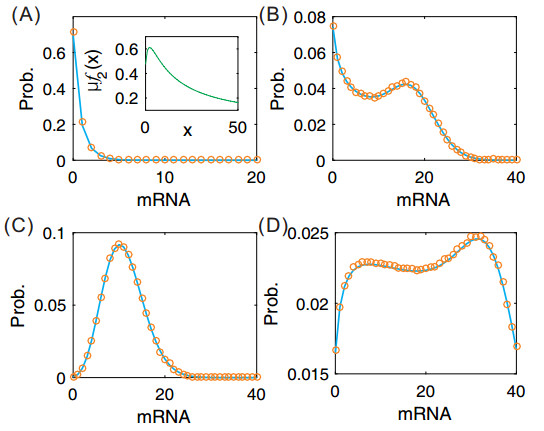
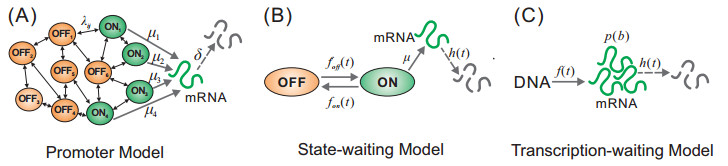
Figures(7)

Jiajun Zhang, Tianshou Zhou. Stationary moments, distribution conjugation and phenotypic regions in stochastic gene transcription[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 6134-6166. doi: 10.3934/mbe.2019307







 DownLoad:
DownLoad: