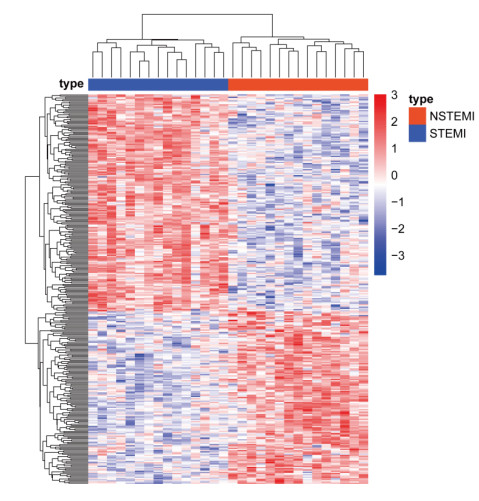
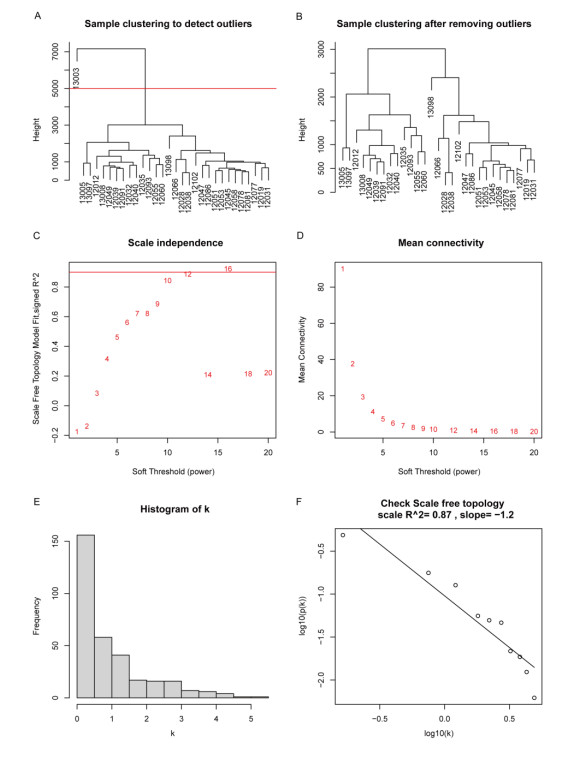
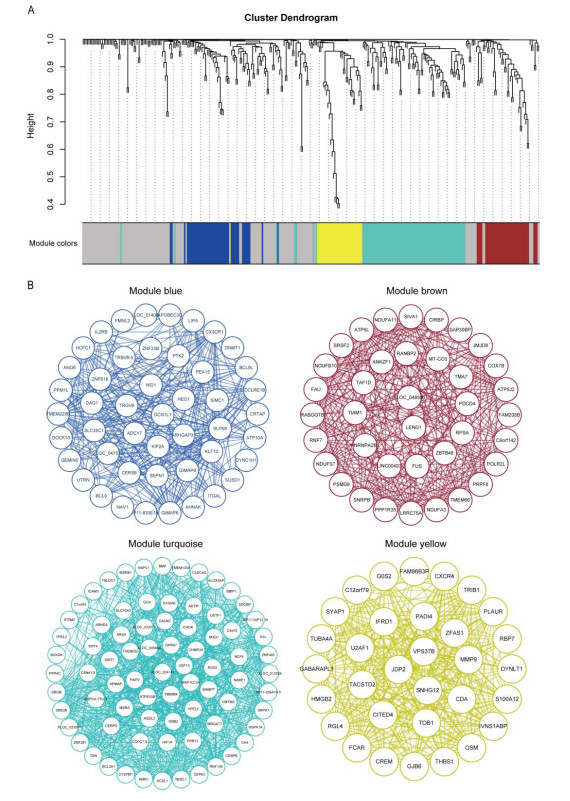
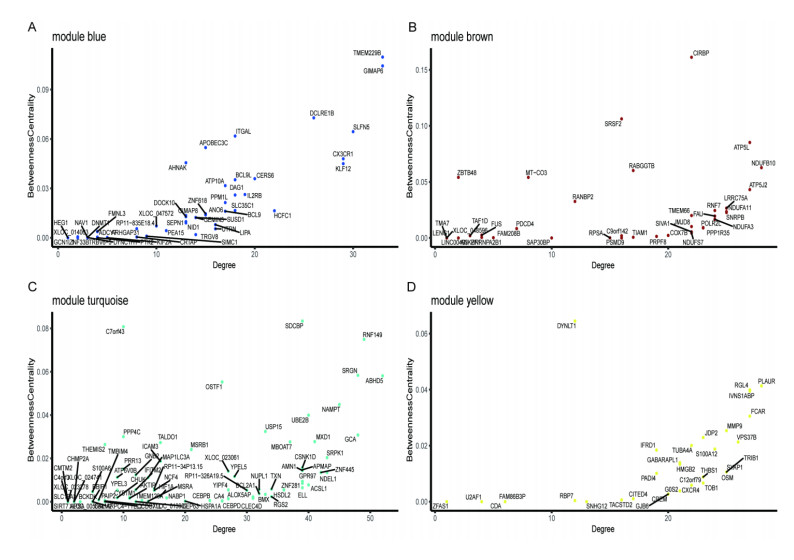
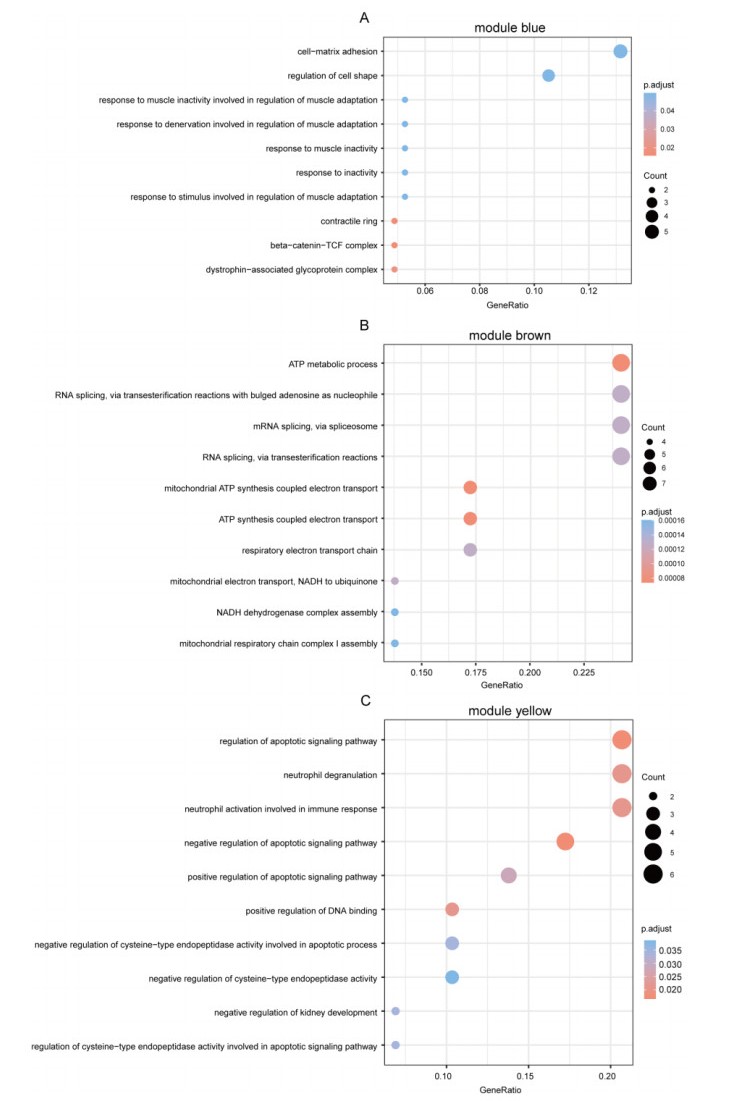
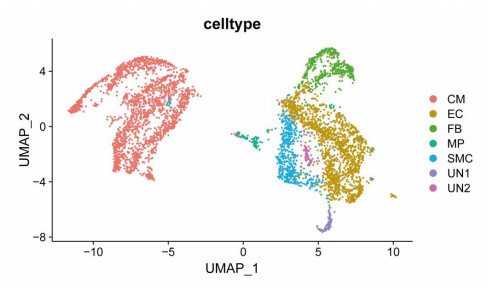
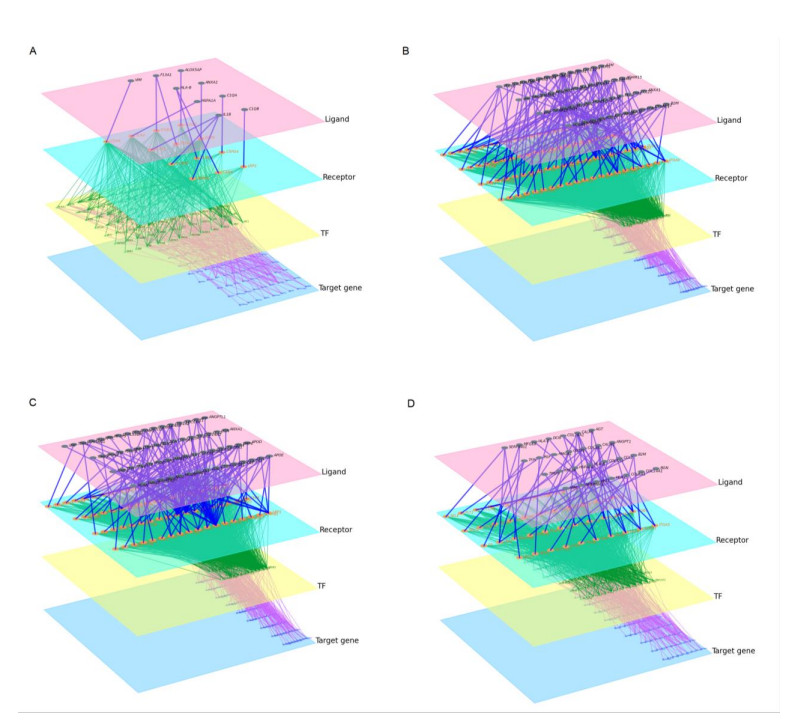
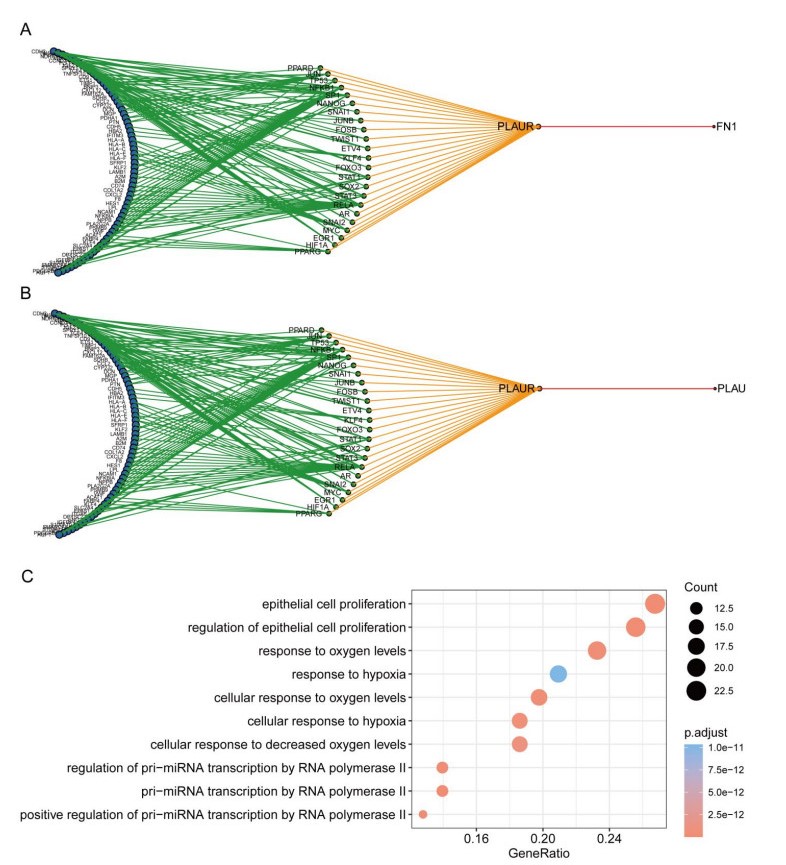
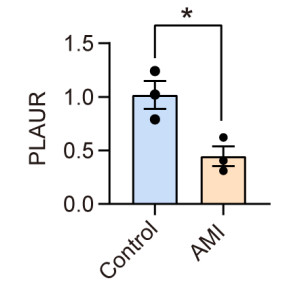
Cardiovascular and cerebrovascular diseases are leading causes of death worldwide, accounting for more than 40% of all deaths in China. Acute myocardial infarction (AMI) is a common cardiovascular disease and traditionally divided into ST-segment (STEMI) and non-ST-segment elevation myocardial infarction (NSTEMI), which are known with different prognoses and treatment strategies. However, key regulatory genes and pathways involved in AMI that may be used as potential biomarker for prognosis are unknown. In this study, we employed both bulk and single-cell RNA-seq to construct gene regulatory networks and cell-cell communication networks. We first constructed weighted gene co-expression networks for differential expressed genes between STEMI and NSTEMI patients based on whole-blood RNA-seq transcriptomics. Network topological attributes (e.g., node degree, betweenness) were analyzed to identify key genes involved in different functional network modules. Furthermore, we used single-cell RNA-seq data to construct multilayer signaling network to infer regulatory mechanisms of the above key genes. PLAUR (receptor for urokinase plasminogen activator) was found to play a vital role in transducing inter-cellular signals from endothelial cells and fibroblast cells to intra-cellular pathways of myocardial cells, leading to gene expression involved in cellular response to hypoxia. Our study sheds lights on identifying molecular biomarkers for diagnosis and prognosis of AMI, and provides candidate key regulatory genes for further experimental validation.
Citation: Jiaxin Luo, Lin Wu, Dinghui Liu, Zhaojun Xiong, Linli Wang, Xiaoxian Qian, Xiaoqiang Sun. Gene regulatory network analysis identifies key genes and regulatory mechanisms involved in acute myocardial infarction using bulk and single cell RNA-seq data[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 7774-7789. doi: 10.3934/mbe.2021386
Cardiovascular and cerebrovascular diseases are leading causes of death worldwide, accounting for more than 40% of all deaths in China. Acute myocardial infarction (AMI) is a common cardiovascular disease and traditionally divided into ST-segment (STEMI) and non-ST-segment elevation myocardial infarction (NSTEMI), which are known with different prognoses and treatment strategies. However, key regulatory genes and pathways involved in AMI that may be used as potential biomarker for prognosis are unknown. In this study, we employed both bulk and single-cell RNA-seq to construct gene regulatory networks and cell-cell communication networks. We first constructed weighted gene co-expression networks for differential expressed genes between STEMI and NSTEMI patients based on whole-blood RNA-seq transcriptomics. Network topological attributes (e.g., node degree, betweenness) were analyzed to identify key genes involved in different functional network modules. Furthermore, we used single-cell RNA-seq data to construct multilayer signaling network to infer regulatory mechanisms of the above key genes. PLAUR (receptor for urokinase plasminogen activator) was found to play a vital role in transducing inter-cellular signals from endothelial cells and fibroblast cells to intra-cellular pathways of myocardial cells, leading to gene expression involved in cellular response to hypoxia. Our study sheds lights on identifying molecular biomarkers for diagnosis and prognosis of AMI, and provides candidate key regulatory genes for further experimental validation.

| [1] | G. Heusch, B. J. Gersh, The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge, Eur. Heart J., 38 (2017), 774-784. |
| [2] | GBD 2017 Disease and Injury Incidence and Prevalence Collaborators, Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017, Lancet., 392 (2018), 1789-1858. |
| [3] |
Y. Wang, J. Li, X. Zheng, Z. Jiang, S. Hu, R. K. Wadhera, et al., Risk Factors Associated With Major Cardiovascular Events 1 Year After Acute Myocardial Infarction, JAMA Netw. Open., 1 (2018), e181079. doi: 10.1001/jamanetworkopen.2018.1079

|
| [4] | X. Sheng, T. Fan, X. Jin, Identification of Key Genes Involved in Acute Myocardial Infarction by Comparative Transcriptome Analysis, Biomed. Res. Int., 2020 (2020), 1470867. |
| [5] |
M. S. Sabatine, PCSK9 inhibitors: clinical evidence and implementation, Nat. Rev. Cardiol., 16 (2019), 155-165. doi: 10.1038/s41569-018-0107-8
|
| [6] |
P. M. Ridker, B. M. Everett, T. Thuren, J. G. MacFadyen, W. H. Chang, C. Ballantyne, et al., Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease, N. Engl. J. Med., 377 (2017), 1119-1131. doi: 10.1056/NEJMoa1707914
|
| [7] |
P. Ramalingam, M. G. Poulos, E. Lazzari, M. C. Gutkin, D. Lopez, C. C. Kloss, et al., Chronic activation of endothelial MAPK disrupts hematopoiesis via NFKB dependent inflammatory stress reversible by SCGF, Nat. Commun., 11 (2020), 666. doi: 10.1038/s41467-020-14478-8
|
| [8] |
C. Lechauve, J. T. Butcher, A. Freiwan, L. A. Biwer, J. M. Keith, M. E. Good, et al., Endothelial cell α-globin and its molecular chaperone α-hemoglobin-stabilizing protein regulate arteriolar contractility, J. Clin. Invest., 128 (2018), 5073-5082. doi: 10.1172/JCI99933
|
| [9] | H. Y. Hwang, J. S. Shim, D. Kim, H. J. Kwon, Antidepressant drug sertraline modulates AMPK-MTOR signaling-mediated autophagy via targeting mitochondrial VDAC1 protein, Autophagy, (2020), 1-17. |
| [10] |
M. Cui, Z. Wang, K. Chen, A. M. Shah, W. Tan, L. Duan, et al., Dynamic Transcriptional Responses to Injury of Regenerative and Non-regenerative Cardiomyocytes Revealed by Single-Nucleus RNA Sequencing, Dev. Cell., 53 (2020), 102-116. doi: 10.1016/j.devcel.2020.02.019
|
| [11] |
Ruiz-Villalba, J. P. Romero, S. C. Hernández, A. Vilas-Zornoza, N. Fortelny, L. Castro-Labrador, et al., Single-Cell RNA Sequencing Analysis Reveals a Crucial Role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) Cardiac Fibroblasts After Myocardial Infarction, Circulation, 142 (2020), 1831-1847. doi: 10.1161/CIRCULATIONAHA.119.044557
|
| [12] |
M. Chiesa, L. Piacentini, E. Bono, V. Milazzo, J. Campodonico, G. Marenzi, et al., Whole blood transcriptome profile at hospital admission discriminates between patients with ST-segment elevation and non-ST-segment elevation acute myocardial infarction, Sci. Rep., 10 (2020), 8731. doi: 10.1038/s41598-020-65527-7
|
| [13] |
D. Risso, J. Ngai, T. P. Speed, S. Dudoit, Normalization of RNA-seq data using factor analysis of control genes or samples, Nat. Biotechnol., 32 (2014), 896-902. doi: 10.1038/nbt.2931
|
| [14] |
M. D. Robinson, D. J. McCarthy, G. K. Smyth, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data, Bioinformatics, 26 (2010), 139-140. doi: 10.1093/bioinformatics/btp616
|
| [15] |
P. Langfelder, S. Horvath, WGCNA: an R package for weighted correlation network analysis, BMC Bioinform., 9 (2008), 559. doi: 10.1186/1471-2105-9-559
|
| [16] | P. Shannon, A. Markiel, O.Ozier, N. S. Baliga, J. T. Wang, D. Ramage, et al., Cytoscape: a sofware environment for integrated models of biomolecular interaction networks, Genome Res., 13 (2013), 2498-2504. |
| [17] |
L. Wang, P. Yu, B. Zhou, J. Song, Z. Li, M. Zhang, et al., Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function, Nat. Cell Biol., 22 (2020), 108-119. doi: 10.1038/s41556-019-0446-7
|
| [18] |
W. V. Li, J. J. Li, An accurate and robust imputation method scImpute for single-cell RNA-seq data, Nat. Commun., 9 (2018), 997. doi: 10.1038/s41467-018-03405-7
|
| [19] |
T. Stuart, A. Butler, P. Hoffman, C. Hafemeister, E. Papalexi, W. M. Mauck, et al., Comprehensive Integration of Single-Cell Data, Cell, 177 (2019), 1888-1902.e21. doi: 10.1016/j.cell.2019.05.031
|
| [20] |
J. Cheng, J. Zhang, Z. Wu, X. Sun, Inferring microenvironmental regulation of gene expression from single-cell RNA sequencing data using scMLnet with an application to COVID-19, Brief Bioinform., 22 (2021), 988-1005. doi: 10.1093/bib/bbaa327
|
| [21] |
G. Yu, L. G. Wang, Y. Han, Q. Y. He, clusterProfiler: an R package for comparing biological themes among gene clusters, OMICS., 16 (2012), 284-287. doi: 10.1089/omi.2011.0118
|
| [22] | J. M. Tonne, T. Sakuma, M. C. Deeds, M. Munoz-Gomez, M. A. Barry, Y. C. Kudva, et al., Global gene expression profiling of pancreatic islets in mice during streptozotocin-induced β-cell damage and pancreatic Glp-1 gene therapy, Dis. Model. Mech., 6 (2013), 1236-1245. |
| [23] |
M Ploug, E Rønne, N Behrendt, A. L. Jensen, F. Blasi, K. Danø, Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol, J. Biol. Chem., 266 (1991), 1926-1933. doi: 10.1016/S0021-9258(18)52382-6
|
| [24] |
C. E. Almasi, L. Drivsholm, H. Pappot, G. Høyer-Hansen, I. J. Christensen, The liberated domain I of urokinase plasminogen activator receptor--a new tumour marker in small cell lung cancer, APMIS., 121 (2013), 189-196. doi: 10.1111/j.1600-0463.2012.02955.x
|
| [25] | R. Riisbro, I. J. Christensen, T. Piironen, M. Greenall, B. Larsen, R. W. Stephens et al., Prognostic significance of soluble urokinase plasminogen activator receptor in serum and cytosol of tumor tissue from patients with primary breast cancer, Clin. Cancer Res., 8 (2002), 1132-1141. |
| [26] |
S. Sharma, P. G. Jackson, J. Makan, Cardiac troponins, J. Clin. Pathol., 57 (2004), 1025-1026. doi: 10.1136/jcp.2003.015420
|
| [27] |
S. Aydin, K. Ugur, S. Aydin, İ. Sahin, M. Yardim, Biomarkers in acute myocardial infarction: current perspectives, Vasc. Health Risk Manag., 15 (2019), 1-10. doi: 10.2147/VHRM.S166157
|
Figures(9) / Tables(1)

Jiaxin Luo, Lin Wu, Dinghui Liu, Zhaojun Xiong, Linli Wang, Xiaoxian Qian, Xiaoqiang Sun. Gene regulatory network analysis identifies key genes and regulatory mechanisms involved in acute myocardial infarction using bulk and single cell RNA-seq data[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 7774-7789. doi: 10.3934/mbe.2021386







 DownLoad:
DownLoad: