Citation: Sonia Shastri, Ravichandra Vemuri, Nuri Gueven, Madhur D. Shastri, Rajaraman Eri. Molecular mechanisms of intestinal inflammation leading to colorectal cancer[J]. AIMS Biophysics, 2017, 4(1): 152-177. doi: 10.3934/biophy.2017.1.152

| [1] |
Cosnes J, Gower-Rousseau C, Seksik P, et al. (2011) Epidemiology and natural history of inflammatory bowel diseases. Gastroenterol 140: 1785–1794. doi: 10.1053/j.gastro.2011.01.055

|
| [2] |
Shanahan F, Bernstein CN (2009) The evolving epidemiology of inflammatory bowel disease. Curr Opin Gastroenterol 25: 301–305. doi: 10.1097/MOG.0b013e32832b12ef
|
| [3] | Economou M, Pappas G (2008) New global map of Crohn's disease: Genetic, environmental, and socioeconomic correlations. Inflamm Bowel Dis 4: 709–720. |
| [4] |
Mulder DJ, Noble AJ, Justinich CJ, et al. (2014) A tale of two diseases: the history of inflammatory bowel disease. J Crohns Colitis 8: 341–348. doi: 10.1016/j.crohns.2013.09.009
|
| [5] | Hanauer SB (2006) Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis 12: S3–S9. |
| [6] |
Dubinsky M (2008) Special issues in pediatric inflammatory bowel disease. World J Gastroenterol 14: 413–420. doi: 10.3748/wjg.14.413
|
| [7] |
Diefenbach KA, Breuer CK (2006) Pediatric inflammatory bowel disease. World J Gastroenterol 12: 3204–3212. doi: 10.3748/wjg.v12.i20.3204
|
| [8] |
Ekbom A, Helmick C, Zack M, et al. (1991) The epidemiology of inflammatory bowel disease: a large, population-based study in Sweden. Gastroenterol 100: 350–358. doi: 10.1016/0016-5085(91)90202-V
|
| [9] |
CROHN BB, Rosenberg H (1925) The sigmoidoscopic picture of chronic ulcerative colitis (non-specific). Am J Med Sci 170: 220–227. doi: 10.1097/00000441-192508010-00006
|
| [10] |
Rutter MD, Saunders BP, Wilkinson KH, et al. (2006) Thirty-year analysis of a colonoscopic surveillance program for neoplasia in ulcerative colitis. Gastroenterol 130: 1030–1038. doi: 10.1053/j.gastro.2005.12.035
|
| [11] |
Beaugerie L, Svrcek M, Seksik P, et al. (2013) Risk of colorectal high-grade dysplasia and cancer in a prospective observational cohort of patients with inflammatory bowel disease. Gastroenterol 145: 166–175. doi: 10.1053/j.gastro.2013.03.044
|
| [12] |
Eaden J, Abrams K, Mayberry J (2001) The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 48: 526–535. doi: 10.1136/gut.48.4.526
|
| [13] |
Canavan C, Abrams K, Mayberry J (2006) Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn's disease. Aliment Pharmacol Ther 23: 1097–1104. doi: 10.1111/j.1365-2036.2006.02854.x
|
| [14] | Itzkowitz SH, Yio X (2004) Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol 287: G7–G17. |
| [15] | Kornfeld D, Ekbom A, Ihre T, et al. (1997) Is there an excess risk for colorectal cancer in patients with ulcerative colitis and concomitant primary sclerosing cholangitis? A population based study. Gut 41: 522–525. |
| [16] |
Matula S, Croog V, Itzkowitz S, et al. (2005) Chemoprevention of colorectal neoplasia in ulcerative colitis: the effect of 6-mercaptopurine. Clin Gastroenterol Hepatol 3: 1015–1021. doi: 10.1016/S1542-3565(05)00738-X
|
| [17] |
Terdiman JP, Steinbuch M, Blumentals WA, et al. (2007) 5-Aminosalicylic acid therapy and the risk of colorectal cancer among patients with inflammatory bowel disease. Inflamm Bowel Dis 13: 367–371. doi: 10.1002/ibd.20074
|
| [18] |
Kiesslich R, Goetz M, Lammersdorf K, et al. (2007) Chromoscopy-guided endomicroscopy increases the diagnostic yield of intraepithelial neoplasia in ulcerative colitis. Gastroenterol 132: 874–882. doi: 10.1053/j.gastro.2007.01.048
|
| [19] |
Schneider MR, Hoeflich A, Fischer JR, et al. (2000) Interleukin-6 stimulates clonogenic growth of primary and metastatic human colon carcinoma cells. Cancer Lett 151: 31–38. doi: 10.1016/S0304-3835(99)00401-2
|
| [20] |
Sakamoto K, Maeda S, Hikiba Y, et al. (2009) Constitutive NF-κB activation in colorectal carcinoma plays a key role in angiogenesis, promoting tumor growth. Clin Cancer Res 15: 2248–2258. doi: 10.1158/1078-0432.CCR-08-1383
|
| [21] |
Kang KA, Zhang R, Kim GY, et al. (2012) Epigenetic changes induced by oxidative stress in colorectal cancer cells: methylation of tumor suppressor RUNX3. Tumor Biol 33: 403–412. doi: 10.1007/s13277-012-0322-6
|
| [22] |
Ning Y, Manegold PC, Hong YK, et al. (2011) Interleukin-8 is associated with proliferation, migration, angiogenesis and chemosensitivity in vitro and in vivo in colon cancer cell line models. Int J Cancer 128: 2038–2049. doi: 10.1002/ijc.25562
|
| [23] |
Huch M, Koo BK (2015) Modeling mouse and human development using organoid cultures. Development 142: 3113–3125. doi: 10.1242/dev.118570
|
| [24] | Van Limbergen J, Geddes K, Henderson P, et al. (2013) Paneth cell marker CD24 in NOD2 knockout organoids and in inflammatory bowel disease (IBD). Gut:gutjnl-2013-305077. |
| [25] |
van de Wetering M, Francies HE, Francis JM, et al. (2015) Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161: 933–945. doi: 10.1016/j.cell.2015.03.053
|
| [26] |
Fatehullah A, Tan SH, Barker N (2016) Organoids as an in vitro model of human development and disease. Nat Cell Biol 18: 246–254. doi: 10.1038/ncb3312
|
| [27] |
Dharmani P, Leung P, Chadee K (2011) Tumor necrosis factor-α and Muc2 mucin play major roles in disease onset and progression in dextran sodium sulphate-induced colitis. PLoS One 6: e25058. doi: 10.1371/journal.pone.0025058
|
| [28] |
Heazlewood CK, Cook MC, Eri R, et al. (2008) Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med 5: e54. doi: 10.1371/journal.pmed.0050054
|
| [29] |
Half E, Bercovich D, Rozen P (2009) Familial adenomatous polyposis. Orphanet J Rare Dis 4: 22. doi: 10.1186/1750-1172-4-22
|
| [30] |
Fodde R, Smits R (2001) Disease model: familial adenomatous polyposis. Trends Mol Med 7: 369–373. doi: 10.1016/S1471-4914(01)02050-0
|
| [31] |
Quesada CF, Kimata H, Mori M, et al. (1998) Piroxicam and acarbose as chemopreventive agents for spontaneous intestinal adenomas in APC gene 1309 knockout mice. JPN J Cancer Res 89: 392–396. doi: 10.1111/j.1349-7006.1998.tb00576.x
|
| [32] | Corpet DE, Pierre F (2003) Point: From animal models to prevention of colon cancer. Systematic review of chemoprevention in min mice and choice of the model system. Cancer Epidemiol Biomarkers Prev 12: 391–400. |
| [33] |
Aoki K, Tamai Y, Horiike S, et al. (2003) Colonic polyposis caused by mTOR-mediated chromosomal instability in Apc+/Δ716 Cdx2+/− compound mutant mice. Nat Genet 35: 323–330. doi: 10.1038/ng1265
|
| [34] |
Heyer J, Yang K, Lipkin M, et al. (1999) Mouse models for colorectal cancer. Oncogene 18: 5325–5333. doi: 10.1038/sj.onc.1203036
|
| [35] |
Velcich A, Yang W, Heyer J, et al. (2002) Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 295: 1726–1729. doi: 10.1126/science.1069094
|
| [36] |
Van der Sluis M, De Koning BA, De Bruijn AC, et al. (2006) Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterol 131: 117–129. doi: 10.1053/j.gastro.2006.04.020
|
| [37] |
Zhu Y, Richardson JA, Parada LF, et al. (1998) Smad3 mutant mice develop metastatic colorectal cancer. Cell 94: 703–714. doi: 10.1016/S0092-8674(00)81730-4
|
| [38] |
Yang X, Letterio JJ, Lechleider RJ, et al. (1999) Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J 18: 1280–1291. doi: 10.1093/emboj/18.5.1280
|
| [39] | Perše M, Cerar A (2012) Dextran sodium sulphate colitis mouse model: traps and tricks. Biomed Res Int 2012. |
| [40] |
Delker DA, McKnight SJ, Rosenberg DW (1998) The role of alcohol dehydrogenase in the metabolism of the colon carcinogen methylazoxymethanol. Toxicol Sci 45: 66–71. doi: 10.1093/toxsci/45.1.66
|
| [41] | Haase P, Cowen D, Knowles J (1973) Histogenesis of colonic tumours in mice induced by dimethyl hydrazine. J Pathol 109: Px. |
| [42] |
Neufert C, Becker C, Neurath MF (2007) An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc 2: 1998–2004. doi: 10.1038/nprot.2007.279
|
| [43] |
Tanaka T, Kohno H, Suzuki R, et al. (2003) A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci 94: 965–973. doi: 10.1111/j.1349-7006.2003.tb01386.x
|
| [44] |
De Robertis M, Massi E, Poeta ML, et al. (2011) The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J Carcinog 10: 9. doi: 10.4103/1477-3163.78279
|
| [45] | Reddy BS, Ohmori T (1981) Effect of intestinal microflora and dietary fat on 3, 2'-dimethyl-4-aminobiphenyl-induced colon carcinogenesis in F344 rats. Cancer Res 41: 1363–1367. |
| [46] |
Hasegawa R, Sano M, Tamano S, et al. (1993) Dose-dependence of 2-amino-1-methy1-6-phen-ylimidazo [4, 5-b]-pyridine (PhIP) carcinogenicity in rats. Carcinogenesis 14: 2553–2557. doi: 10.1093/carcin/14.12.2553
|
| [47] |
Wanibuchi H, Salim EI, Morimura K, et al. (2005) Lack of large intestinal carcinogenicity of 2-amino-1-methyl-6-phenylimidazo [4, 5-b] pyridine at low doses in rats initiated with azoxymethane. Int J Cancer 115: 870–878. doi: 10.1002/ijc.20960
|
| [48] | Kobaek-Larsen M, Thorup I, Diederichsen A, et al. (2000) Review of colorectal cancer and its metastases in rodent models: comparative aspects with those in humans. Comp Med 50: 16–26. |
| [49] |
Narisawa T, Magadia NE, Weisburger JH, et al. (1974) Promoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N-Methyl-N' nitro-N-nitrosoguanidine in Rats. J Natl Cancer Inst 53: 1093–1097. doi: 10.1093/jnci/53.4.1093
|
| [50] |
Einerhand AW, Renes IB, Makkink MK, et al. (2002) Role of mucins in inflammatory bowel disease: important lessons from experimental models. Eur J GastroenterolHepatol 14: 757–765. doi: 10.1097/00042737-200207000-00008
|
| [51] |
Randall-Demllo S, Fernando R, Brain T, et al. (2016) Characterisation of colonic dysplasia-like epithelial atypia in murine colitis. World J Gastroenterol 22: 8334–8348. doi: 10.3748/wjg.v22.i37.8334
|
| [52] |
Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12: 931–947. doi: 10.1038/nrd4002
|
| [53] |
Jackson AL, Loeb LA (2001) The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mut Res Fund Mol Mech Mutagen 477: 7–21. doi: 10.1016/S0027-5107(01)00091-4
|
| [54] | Kawanishi S, Hiraku Y, Pinlaor S, et al. (2006) Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem 387: 365–372. |
| [55] |
Tüzün A, Erdil A, İnal V, et al. (2002) Oxidative stress and antioxidant capacity in patients with inflammatory bowel disease. Clin Biochem 35: 569–572. doi: 10.1016/S0009-9120(02)00361-2
|
| [56] |
Nair J, Gansauge F, Beger H, et al. (2006) Increased etheno-DNA adducts in affected tissues of patients suffering from Crohn's disease, ulcerative colitis, and chronic pancreatitis. Antioxid Redox Signal 8: 1003–1010. doi: 10.1089/ars.2006.8.1003
|
| [57] |
Vong LB, Yoshitomi T, Matsui H, et al. (2015) Development of an oral nanotherapeutics using redox nanoparticles for treatment of colitis-associated colon cancer. Biomaterials 55: 54–63. doi: 10.1016/j.biomaterials.2015.03.037
|
| [58] | Solomon H, Brosh R, Buganim Y, et al. (2010) Inactivation of the p53 tumor suppressor gene and activation of the Ras oncogene: cooperative events in tumorigenesis. Discov Med 9: 448–454. |
| [59] | Huang H, Wang H, Lloyd RS, et al. (2008) Conformational interconversion of the trans-4-hydroxynonenal-derived (6S, 8R, 11S) 1, N 2-deoxyguanosine adduct when mismatched with deoxyadenosine in DNA. Chem Res Toxicol 22: 187–200. |
| [60] |
Barrett CW, Ning W, Chen X, et al. (2013) Tumor suppressor function of the plasma glutathione peroxidase gpx3 in colitis-associated carcinoma. Cancer Res 73: 1245–1255. doi: 10.1158/0008-5472.CAN-12-3150
|
| [61] |
Curtin NJ (2012) DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 12: 801–817. doi: 10.1038/nrc3399
|
| [62] |
Khor TO, Huang MT, Prawan A, et al. (2008) Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res 1: 187–191. doi: 10.1158/1940-6207.CAPR-08-0028
|
| [63] | Meira LB, Bugni JM, Green SL, et al. (2008) DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest 118: 2516–2525. |
| [64] |
Sohn JJ, Schetter AJ, Yfantis HG, et al. (2012) Macrophages, nitric oxide and microRNAs are associated with DNA damage response pathway and senescence in inflammatory bowel disease. PLoS One 7: e44156. doi: 10.1371/journal.pone.0044156
|
| [65] | Kohonen-Corish MR, Daniel JJ, te Riele H, et al. (2002) Susceptibility of Msh2-deficient mice to inflammation-associated colorectal tumors. Cancer Res 62: 2092–2097. |
| [66] | Fleisher AS, Esteller M, Harpaz N, et al. (2000) Microsatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1. Cancer Res 60: 4864–4868. |
| [67] |
Redston MS, Papadopoulos N, Caldas C, et al. (1995) Common occurrence of APC and K-ras gene mutations in the spectrum of colitis-associated neoplasias. Gastroenterol 108: 383–392. doi: 10.1016/0016-5085(95)90064-0
|
| [68] |
Burmer GC, Rabinovitch PS, Haggitt RC, et al. (1992) Neoplastic progression in ulcerative colitis: histology, DNA content, and loss of a p53 allele. Gastroenterol 103: 1602–1610. doi: 10.1016/0016-5085(92)91184-6
|
| [69] |
Yashiro M (2015) Molecular alterations of colorectal cancer with inflammatory bowel disease. Dig Dis Sci 60: 2251–2263. doi: 10.1007/s10620-015-3646-4
|
| [70] |
Mikami T, Yoshida T, Numata Y, et al. (2007) Low frequency of promoter methylation of O6-Methylguanine DNA methyltransferase and hMLH1 in ulcerative colitis-associated tumors. Am J Clin Pathol 127: 366–373. doi: 10.1309/RFETXN6387KLQ1LD
|
| [71] |
Foran E, Garrity-Park MM, Mureau C, et al. (2010) Upregulation of DNA methyltransferase-mediated gene silencing, anchorage-independent growth, and migration of colon cancer cells by interleukin-6. Mol Cancer Res 8: 471–481. doi: 10.1158/1541-7786.MCR-09-0496
|
| [72] | Hartnett L, Egan LJ (2012) Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis bgs006. |
| [73] |
Nakazawa T, Kondo T, Ma D, et al. (2012) Global histone modification of histone H3 in colorectal cancer and its precursor lesions. Hum Pathol 43: 834–842. doi: 10.1016/j.humpath.2011.07.009
|
| [74] |
Li Q, Chen H (2012) Silencing of Wnt5a during colon cancer metastasis involves histone modifications. Epigenetics 7: 551–558. doi: 10.4161/epi.20050
|
| [75] |
Binder H, Steiner L, Przybilla J, et al. (2013) Transcriptional regulation by histone modifications: towards a theory of chromatin re-organization during stem cell differentiation. Phys Biol 10: 026006. doi: 10.1088/1478-3975/10/2/026006
|
| [76] |
Bardhan K, Liu K (2013) Epigenetics and colorectal cancer pathogenesis. Cancers 5: 676–713. doi: 10.3390/cancers5020676
|
| [77] |
Klose RJ, Zhang Y (2007) Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol 8: 307–318. doi: 10.1038/nrm2143
|
| [78] |
Wong JJL, Hawkins NJ, Ward RL (2007) Colorectal cancer: a model for epigenetic tumorigenesis. Gut 56: 140–148. doi: 10.1136/gut.2005.088799
|
| [79] |
Portela A, Esteller M (2010) Epigenetic modifications and human disease. Nat Biotechnol 28: 1057–1068. doi: 10.1038/nbt.1685
|
| [80] |
Glauben R, Batra A, Fedke I, et al. (2006) Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol 176: 5015–5022. doi: 10.4049/jimmunol.176.8.5015
|
| [81] |
Glauben R, Batra A, Stroh T, et al. (2008) Histone deacetylases: novel targets for prevention of colitis-associated cancer in mice. Gut 57: 613–622. doi: 10.1136/gut.2007.134650
|
| [82] |
Griffiths-Jones S, Grocock RJ, Van Dongen S, et al. (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34: D140–D144. doi: 10.1093/nar/gkj112
|
| [83] |
Wu F, Zikusoka M, Trindade A, et al. (2008) MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2α. Gastroenterol 135: 1624–1635. doi: 10.1053/j.gastro.2008.07.068
|
| [84] |
Olaru AV, Selaru FM, Mori Y, et al. (2011) Dynamic changes in the expression of MicroRNA-31 during inflammatory bowel disease-associated neoplastic transformation. Inflamm Bowel Dis 17: 221–231. doi: 10.1002/ibd.21359
|
| [85] | Shi C, Yang Y, Xia Y, et al. (2015) Novel evidence for an oncogenic role of microRNA-21 in colitis-associated colorectal cancer. Gut 308–455. |
| [86] | Svrcek M, El-Murr N, Wanherdrick K, et al. (2013) Overexpression of microRNAs-155 and 21 targeting mismatch repair proteins in inflammatory bowel diseases. Carcinogenesis bgs408. |
| [87] |
Polytarchou C, Hommes DW, Palumbo T, et al. (2015) MicroRNA214 is associated with progression of ulcerative colitis, and inhibition reduces development of colitis and colitis-associated cancer in mice. Gastroenterol 149: 981–992. doi: 10.1053/j.gastro.2015.05.057
|
| [88] | Cristóbal I, Manso R, Gónzález-Alonso P, et al. (2015) Clinical value of miR-26b discriminating ulcerative colitis-associated colorectal cancer in the subgroup of patients with metastatic disease. Inflamm Bowel Dis 21: E24–E25. |
| [89] |
Ludwig K, Fassan M, Mescoli C, et al. (2013) PDCD4/miR-21 dysregulation in inflammatory bowel disease-associated carcinogenesis. Virchows Arch 462: 57–63. doi: 10.1007/s00428-012-1345-5
|
| [90] |
Yang L, Belaguli N, Berger DH (2009) MicroRNA and colorectal cancer. World J Surg 33: 638–646. doi: 10.1007/s00268-008-9865-5
|
| [91] |
Kanaan Z, Rai SN, Eichenberger MR, et al. (2012) Differential MicroRNA expression tracks neoplastic progression in inflammatory bowel disease-associated colorectal cancer. Hum Mutat 33: 551–560. doi: 10.1002/humu.22021
|
| [92] |
Feng R, Chen X, Yu Y, et al. (2010) miR-126 functions as a tumour suppressor in human gastric cancer. Cancer Lett 298: 50–63. doi: 10.1016/j.canlet.2010.06.004
|
| [93] |
Fasseu M, Tréton X, Guichard C, et al. (2010) Identification of restricted subsets of mature microRNA abnormally expressed in inactive colonic mucosa of patients with inflammatory bowel disease. PloS One 5: e13160. doi: 10.1371/journal.pone.0013160
|
| [94] | Wang W, Li X, Zheng D, et al. (2015) Dynamic changes and functions of macrophages and M1/M2 subpopulations during ulcerative colitis-associated carcinogenesis in an AOM/DSS mouse model. Mol Med Rep 11: 2397–2406. |
| [95] |
Francescone R, Hou V, Grivennikov SI (2015) Cytokines, IBD, and colitis-associated cancer. Inflamm Bowel Dis 21: 409–418. doi: 10.1097/MIB.0000000000000236
|
| [96] |
Sarra M, Pallone F, MacDonald TT, et al. (2010) IL-23/IL-17 axis in IBD. Inflamm Bowel Dis 16: 1808–1813. doi: 10.1002/ibd.21248
|
| [97] | Reinecker HC, Steffen M, Witthoeft T, et al. (1993) Enhand secretion of tumour necrosis factor-alpha, IL-6, and IL-1β by isolated lamina ropria monouclear cells from patients with ulcretive cilitis and Crohn's disease. Clin Exp Immunol 94: 174–181. |
| [98] | Banks C, Bateman A, Payne R, et al. (2003) Chemokine expression in IBD. Mucosal chemokine expression is unselectively increased in both ulcerative colitis and Crohn's disease. J Pathol 199: 28–35. |
| [99] |
Wiercinska-Drapalo A, Flisiak R, Jaroszewicz J, et al. (2005) Plasma interleukin-18 reflects severity of ulcerative colitis. World J Gastroenterol 11: 605–608. doi: 10.3748/wjg.v11.i4.605
|
| [100] |
Bisping G, Lügering N, Lütke-Brintrup S, et al. (2001) Patients with inflammatory bowel disease (IBD) reveal increased induction capacity of intracellular interferon-gamma (IFN-γ) in peripheral CD8+ lymphocytes co-cultured with intestinal epithelial cells. Clin Exp Immunol 123: 15–22. doi: 10.1046/j.1365-2249.2001.01443.x
|
| [101] | Hyun YS, Han DS, Lee AR, et al. (2012) Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis bgs106. |
| [102] |
Onizawa M, Nagaishi T, Kanai T, et al. (2009) Signaling pathway via TNF-α/NF-κB in intestinal epithelial cells may be directly involved in colitis-associated carcinogenesis. Am J Physiol Gastrointest Liver Physiol 296: G850–G859. doi: 10.1152/ajpgi.00071.2008
|
| [103] |
Matsumoto S, Hara T, Mitsuyama K, et al. (2010) Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble–IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J Immunol 184: 1543–1551. doi: 10.4049/jimmunol.0801217
|
| [104] |
Atreya R, Mudter J, Finotto S, et al. (2000) Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med 6: 583–588. doi: 10.1038/75068
|
| [105] | Popivanova BK, Kitamura K, Wu Y, et al. (2008) Blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest 118: 560–570. |
| [106] |
Fukata M, Chen A, Vamadevan AS, et al. (2007) Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterol 133: 1869–1869. doi: 10.1053/j.gastro.2007.09.008
|
| [107] |
Garrett WS, Punit S, Gallini CA, et al. (2009) Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell 16: 208–219. doi: 10.1016/j.ccr.2009.07.015
|
| [108] |
Allen IC, TeKippe EM, Woodford RMT, et al. (2010) The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 207: 1045–1056. doi: 10.1084/jem.20100050
|
| [109] |
Allen IC, Wilson JE, Schneider M, et al. (2012) NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 36: 742–754. doi: 10.1016/j.immuni.2012.03.012
|
| [110] |
Chen GY, Liu M, Wang F, et al. (2011) A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol 186: 7187–7194. doi: 10.4049/jimmunol.1100412
|
| [111] | Eckmann L, Greten T (2004) IKKbeta links inflam. mation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118: 285–296. |
| [112] |
Cooks T, Pateras IS, Tarcic O, et al. (2013) Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 23: 634–646. doi: 10.1016/j.ccr.2013.03.022
|
| [113] |
Grivennikov S, Karin E, Terzic J, et al. (2009) IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15: 103–113. doi: 10.1016/j.ccr.2009.01.001
|
| [114] |
Bollrath J, Phesse TJ, von Burstin VA, et al. (2009) gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 15: 91–102. doi: 10.1016/j.ccr.2009.01.002
|
| [115] |
Ghosh S, Karin M (2002) Missing pieces in the NF-κB puzzle. Cell 109: S81–S96. doi: 10.1016/S0092-8674(02)00703-1
|
| [116] |
Karin M (2006) Nuclear factor-κB in cancer development and progression. Nature 441: 431–436. doi: 10.1038/nature04870
|
| [117] |
Greten FR, Eckmann L, Greten TF, et al. (2004) IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118: 285–296. doi: 10.1016/j.cell.2004.07.013
|
| [118] |
Rose-John S (2012) IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci 8: 1237–1247. doi: 10.7150/ijbs.4989
|
| [119] |
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9: 798–809. doi: 10.1038/nrc2734
|
| [120] |
Pickert G, Neufert C, Leppkes M, et al. (2009) STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 206: 1465–1472. doi: 10.1084/jem.20082683
|
| [121] |
Putoczki TL, Thiem S, Loving A, et al. (2013) Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 24: 257–271. doi: 10.1016/j.ccr.2013.06.017
|
| [122] | Chichlowski M, Sharp JM, Vanderford DA, et al. (2008) Helicobacter typhlonius and Helicobacter rodentium differentially affect the severity of colon inflammation and inflammation-associated neoplasia in IL10-deficient mice. Comp Med 58: 534–541. |
| [123] |
Uronis JM, Mühlbauer M, Herfarth HH, et al. (2009) Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PloS One 4: e6026. doi: 10.1371/journal.pone.0006026
|
| [124] |
O'mahony L, Feeney M, O'halloran S, et al. (2001) Probiotic impact on microbial flora, inflammation and tumour development in IL-10 knockout mice. Aliment Pharmacol Ther 15: 1219–1225. doi: 10.1046/j.1365-2036.2001.01027.x
|
| [125] |
Tözün N, Vardareli E (2016) Gut microbiome and gastrointestinal cancer: les liaisons dangereuses. J Clin Gastroenterol 50: S191–S196. doi: 10.1097/MCG.0000000000000714
|
| [126] | Yamamoto M, Matsumoto S (2016) Gut microbiota and colorectal cancer. Genes and Environ 38: 1–7. |
| [127] |
Abreu MT (2010) Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol 10: 131–144. doi: 10.1038/nri2707
|
| [128] | Grivennikov SI (2013) Inflammation and colorectal cancer: colitis-associated neoplasia, In: Seminars in immunopathology, Springer-Verlag, 229–244. |
| [129] |
Lowe EL, Crother TR, Rabizadeh S, et al. (2010) Toll-like receptor 2 signaling protects mice from tumor development in a mouse model of colitis-induced cancer. PloS One 5: e13027. doi: 10.1371/journal.pone.0013027
|
| [130] |
Fukata M, Chen A, Vamadevan AS, et al. (2007) Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterol 133: 1869–1869. doi: 10.1053/j.gastro.2007.09.008
|
| [131] | Araki T, Toiyama Y, Okita Y, et al. (2016) Surgical treatment for ulcerative colitis-associated cancer or dysplasia, In: Colitis-associated cancer, Springer-Verlag, 109–130. |
| [132] |
Nio K, Higashi D, Kumagai H, et al. (2016) Efficacy and safety analysis of chemotherapy for advanced colitis-associated colorectal cancer in Japan. Anticancer Drugs 27: 457–463. doi: 10.1097/CAD.0000000000000338
|
| [133] |
Impellizzeri D, Esposito E, Mazzon E, et al. (2011) Oleuropein aglycone, an olive oil compound, ameliorates development of arthritis caused by injection of collagen type II in mice. J Pharmacol Exp Ther 339: 859–869. doi: 10.1124/jpet.111.182808
|
| [134] |
Giner E, Recio MC, Ríos JL, et al. (2013) Oleuropein protects against dextran sodium sulfate-induced chronic colitis in mice. J Nat Prod 76: 1113–1120. doi: 10.1021/np400175b
|
| [135] | Acquaviva R, Di Giacomo C, Sorrenti V, et al. (2012) Antiproliferative effect of oleuropein in prostate cell lines. Int J Oncol 41: 31. |
| [136] |
Elamin MH, Daghestani MH, Omer SA, et al. (2013) Olive oil oleuropein has anti-breast cancer properties with higher efficiency on ER-negative cells. Food Chem Toxicol 53: 310–316. doi: 10.1016/j.fct.2012.12.009
|
| [137] |
Giner E, Recio MC, Ríos JL, et al. (2016) Chemopreventive effect of oleuropein in colitis-associated colorectal cancer in c57bl/6 mice. Mol Nutr Food Res 60: 242–255. doi: 10.1002/mnfr.201500605
|
| [138] |
Zhang M, Viennois E, Prasad M, et al. (2016) Edible ginger-derived nanoparticles: a novel therapeutic approach for the prevention and treatment of inflammatory bowel disease and colitis-associated cancer. Biomaterials 101: 321–340. doi: 10.1016/j.biomaterials.2016.06.018
|
| [139] | Lin L, Sun Y, Wang D, et al. (2015) Celastrol ameliorates ulcerative colitis-related colorectal cancer in mice via suppressing inflammatory responses and epithelial-mesenchymal transition. Front Pharmacol 6. |
| [140] |
Shaker ME, Ashamallah SA, Houssen ME (2014) Celastrol ameliorates murine colitis via modulating oxidative stress, inflammatory cytokines and intestinal homeostasis. Chem Biol Interact 210: 26–33. doi: 10.1016/j.cbi.2013.12.007
|
| [141] |
Fung KY, Cosgrove L, Lockett T, et al. (2012) A review of the potential mechanisms for the lowering of colorectal oncogenesis by butyrate. Br J Nutr 108: 820–831. doi: 10.1017/S0007114512001948
|
| [142] |
Hu Y, Le Leu RK, Christophersen CT, et al. (2016) Manipulation of the gut microbiota using resistant starch is associated with protection against colitis-associated colorectal cancer in rats. Carcinogenesis 37: 366–375. doi: 10.1093/carcin/bgw019
|
| [143] | Kong ZL, Kao NJ, Hu JY, et al. (2016) Fucoxanthin-rich brown algae extract decreases inflammation and attenuates colitis-associated colon cancer in mice. J Food Nutr Res 4: 137–147. |
| [144] |
Pandurangan AK, Saadatdoust Z, Hamzah H, et al. (2015) Dietary cocoa protects against colitis-associated cancer by activating the Nrf2/Keap1 pathway. Biofactors 41: 1–14. doi: 10.1002/biof.1195
|
| [145] | Wu WT, Tsai YT, Chen HL (2016) Konjac glucomannan and inulin oligosaccharide attenuated the progression of colitic-associated colon carcinogenesis and modulated immune response in mice. FASEB J 30: 1174. |
| [146] | Periasamy S, Liu CT, Wu WH, et al. (2015) Dietary Ziziphus jujuba fruit influence on aberrant crypt formation and blood cells in colitis-associated colorectal cancer in mice. Asian Pac J Cancer Prev:16: 7561–7566. |
| [147] |
Viennois E, Xiao B, Ayyadurai S, et al. (2014) Micheliolide, a new sesquiterpene lactone that inhibits intestinal inflammation and colitis-associated cancer. Lab Invest 94: 950–965. doi: 10.1038/labinvest.2014.89
|
| [148] | Kunchari Kalaimathi S, Sudhandiran G (2016) Fisetin ameolirates the azoxymethane and dextran sodium sulfate induced colitis associated colorectal cancer. Int J Pharm Clin Res 8: 551–560. |
| [149] |
Yasui Y, Hosokawa M, Mikami N, et al. (2011) Dietary astaxanthin inhibits colitis and colitis-associated colon carcinogenesis in mice via modulation of the inflammatory cytokines. Chem Biol Interact 193: 79–87. doi: 10.1016/j.cbi.2011.05.006
|
| [150] | Yang X, Zhang F, Wang Y, et al. (2013) Oroxylin A inhibits colitis-associated carcinogenesis through modulating the IL-6/STAT3 signaling pathway. Inflamm Bowel Dis 19: 1990–2000. |
| [151] |
Kannengiesser K, Maaser C, Heidemann J, et al. (2008) Melanocortin-derived tripeptide KPV has anti-inflammatory potential in murine models of inflammatory bowel disease. Inflamm Bowel Dis 14: 324–331. doi: 10.1002/ibd.20334
|
| [152] |
Viennois E, Ingersoll SA, Ayyadurai S, et al. (2016) Critical role of PepT1 in promoting colitis-associated cancer and therapeutic benefits of the anti-inflammatory PepT1-mediated tripeptide KPV in a murine model. CMGH Cell Mol Gastroenterol Hepatol 2: 340–357. doi: 10.1016/j.jcmgh.2016.01.006
|
| [153] |
Seraj MJ, Umemoto A, Kajikawa A, et al. (1997) Effects of dietary bile acids on formation of azoxymethane-induced aberrant crypt foci in F344 rats. Cancer Lett 115: 97–103. doi: 10.1016/S0304-3835(97)04719-8
|
| [154] |
Tung BY, Emond MJ, Haggitt RC, et al. (2001) Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med 134: 89–95. doi: 10.7326/0003-4819-134-2-200101160-00008
|
| [155] |
Xie L, Jiang FC, Zhang LM, et al. (2016) Targeting of MyD88 homodimerization by novel synthetic inhibitor TJ-M2010-5 in preventing colitis-associated colorectal cancer. J Natl Cancer Inst 108: djv364. doi: 10.1093/jnci/djv364
|
| [156] |
Amini-Khoei H, Momeny M, Abdollahi A, et al. (2016) Tropisetron suppresses colitis-associated cancer in a mouse model in the remission stage. Int Immunopharmacol 36: 9–16. doi: 10.1016/j.intimp.2016.04.014
|
| [157] |
Drechsler S, Bruntsch U, Eggert J, et al. (1997) Comparison of three tropisetron-containing antiemetic regimens in the prophylaxis of acute and delayed chemotherapy-induced emesis and nausea. Support Care Cancer 5: 387–395. doi: 10.1007/s005200050097
|
| [158] |
Koh SJ, Kim JM, Kim I-K, et al. (2011) Fluoxetine inhibits NF-κB signaling in intestinal epithelial cells and ameliorates experimental colitis and colitis-associated colon cancer in mice. Am J Physiol Gastrointest Liver Physiol 301: G9–G19. doi: 10.1152/ajpgi.00267.2010
|
| [159] |
Tanaka T, Kochi T, Shirakami Y, et al. (2016) Cimetidine and clobenpropit attenuate inflammation-associated colorectal carcinogenesis in male ICR mice. Cancers 8: 25. doi: 10.3390/cancers8020025
|
| [160] |
Masini E, Fabbroni V, Giannini L, et al. (2005) Histamine and histidine decarboxylase up-regulation in colorectal cancer: correlation with tumor stage. Inflamm Res 54: S80–S81. doi: 10.1007/s00011-004-0437-3
|
| [161] |
Miyamoto S, Epifano F, Curini M, et al. (2008) A novel prodrug of 4'-geranyloxy-ferulic acid suppresses colitis-related colon carcinogenesis in mice. Nutr Cancer 60: 675–684. doi: 10.1080/01635580802008286
|
| [162] |
Yao J, Xie J, Xie B, et al. (2016) Therapeutic effect of hydroxychloroquine on colorectal carcinogenesis in experimental murine colitis. Biochem Pharmacol 115: 51–63. doi: 10.1016/j.bcp.2016.06.004
|
| [163] |
Dai Y, Jiao H, Teng G, et al. (2014) Embelin reduces colitis-associated tumorigenesis through limiting IL-6/STAT3 signaling. Mol Cancer Ther 13: 1206–1216. doi: 10.1158/1535-7163.MCT-13-0378
|
| [164] |
Liang J, Nagahashi M, Kim EY, et al. (2013) Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 23: 107–120. doi: 10.1016/j.ccr.2012.11.013
|
| [165] | Kawamori T, Kaneshiro T, Okumura M, et al. (2009) Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J 23: 405–414. |
| [166] |
Snider AJ, Kawamori T, Bradshaw SG, et al. (2009) A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J 23: 143–152. doi: 10.1096/fj.08-118109
|
| [167] |
Wang D, DuBois RN (2010) The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 29: 781–788. doi: 10.1038/onc.2009.421
|
| [168] |
Kohno H, Suzuki R, Sugie S, et al. (2005) Suppression of colitis-related mouse colon carcinogenesis by a COX-2 inhibitor and PPAR ligands. BMC Cancer 5: 1. doi: 10.1186/1471-2407-5-1
|
| [169] |
Setia S, Nehru B, Sanyal SN (2014) The PI3K/Akt pathway in colitis associated colon cancer and its chemoprevention with celecoxib, a Cox-2 selective inhibitor. Biomed Pharmacother 68: 721–727. doi: 10.1016/j.biopha.2014.07.006
|
| [170] |
Glauben R, Sonnenberg E, Zeitz M, et al. (2009) HDAC inhibitors in models of inflammation-related tumorigenesis. Cancer Lett 280: 154–159. doi: 10.1016/j.canlet.2008.11.019
|
| [171] | Wei TT, Lin YT, Tseng RY, et al. (2016) Prevention of colitis and colitis-associated colorectal cancer by a novel polypharmacological Histone deacetylase inhibitor. Am Assoc Cancer Res 22: 4158–4169. |
| [172] |
Reinhard A, Bressenot A, Dassonneville R, et al. (2015) Photodynamic therapy relieves colitis and prevents colitis-associated carcinogenesis in mice. Inflamm Bowel Dis 21: 985–995. doi: 10.1097/MIB.0000000000000354
|
| [173] |
Zhang D, Mi M, Jiang F, et al. (2015) Apple polysaccharide reduces NF-kb mediated colitis-associated colon carcinogenesis. Nutr Cancer 67: 177–190. doi: 10.1080/01635581.2015.965336
|
| [174] |
Yang Y, Cai X, Yang J, et al. (2014) Chemoprevention of dietary digitoflavone on colitis-associated colon tumorigenesis through inducing Nrf2 signaling pathway and inhibition of inflammation. Mol Cancer 13: 48. doi: 10.1186/1476-4598-13-48
|
| [175] | Tian Y, Wang K, Wang Z, et al. (2013) Chemopreventive effect of dietary glutamine on colitis-associated colon tumorigenesis in mice. Carcinogenesis bgt088. |
| [176] |
Tian Y, Wang K, Fan Y, et al. (2016) Chemopreventive effect of dietary glutamineon colitis-associated colorectal cancer is associated with modulation of the DEPTOR/mTOR signaling pathway. Nutrients 8: 261. doi: 10.3390/nu8050261
|
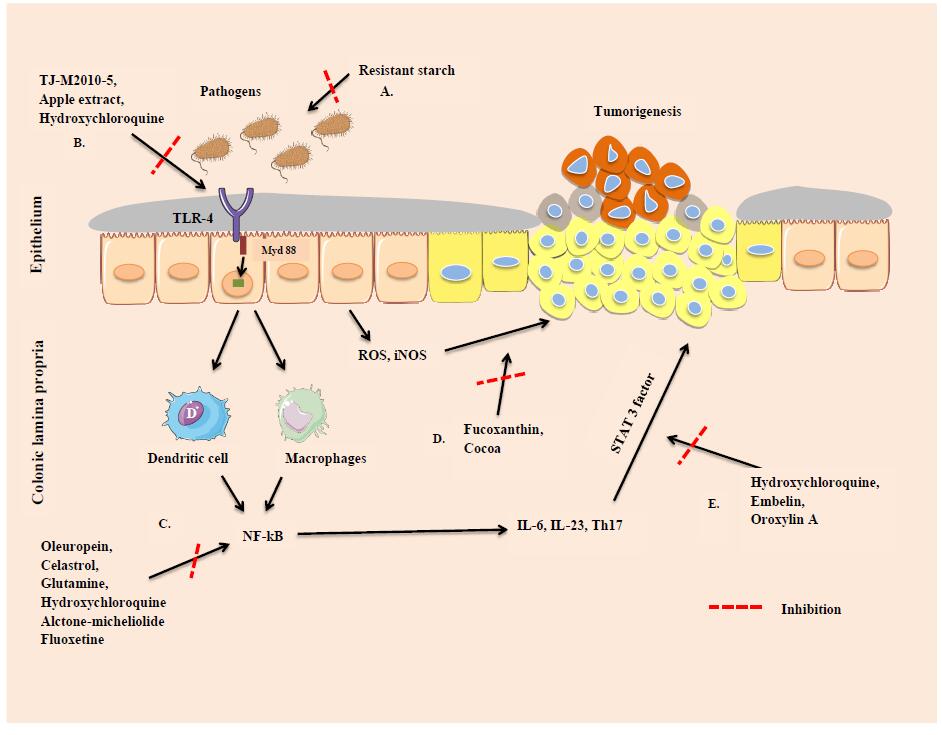
Figures(1) / Tables(1)

Sonia Shastri, Ravichandra Vemuri, Nuri Gueven, Madhur D. Shastri, Rajaraman Eri. Molecular mechanisms of intestinal inflammation leading to colorectal cancer[J]. AIMS Biophysics, 2017, 4(1): 152-177. doi: 10.3934/biophy.2017.1.152







 DownLoad:
DownLoad: