Citation: Masahiro Seike. Histamine suppresses T helper 17 responses mediated by transforming growth factor-β1 in murine chronic allergic contact dermatitis[J]. AIMS Allergy and Immunology, 2018, 2(4): 180-189. doi: 10.3934/Allergy.2018.4.180

| [1] | Kim H, Kim JR, Kang H, et al. (2014) 7,8,4'-Trihyroxyisoflavone attenuates DNCB-induced atopic dermatitis-like symptoms in NC/Nga mice. PLoS One 29: e104938. |
| [2] |
Kitagaki H, Fujisawa S, Watanabe K, et al. (1995) Immediate-type hypersensitivity response followed by a late reaction is induced by repeated epicutaneous application of contact sensitizing agents in mice. J Invest Dermatol 105: 749–755. doi: 10.1111/1523-1747.ep12325538

|
| [3] | Yamaura K, Shimada M, Ueno K (2011) Anthocyanins from bilberry (Vaccinium myrtillus L.) alleviate pruritus in a mouse model of chronic allergic contact dermatitis. Pharmacognosy Res 3: 173–177. |
| [4] |
Boothe WD, Tarbox JA, Tarbox MB (2017) Atopic dermatitis: Pathophysiology. Adv Exp Med Biol 1027: 21–37. doi: 10.1007/978-3-319-64804-0_3
|
| [5] |
Lin L, Xie M, Chen X, et al. (2018) Toll-like receptor 4 attenuates a murine model of atopic dermatitis through inhibition of langerin-positive DCs migration. Exp Dermatol 27: 1015–1022. doi: 10.1111/exd.13698
|
| [6] |
Liu L, Guo D, Liang Q, et al. (2015) The efficacy of sublingual immunotherapy with Dermatophagoides farinae vaccine in a murine atopic dermatitis model. Clin Exp Allergy 45: 815–822. doi: 10.1111/cea.12417
|
| [7] |
Stander S, Steinhoff M (2002) Pathophysiology of pruritus in atopic dermatitis: An overview. Exp Dermatol 11: 12–24. doi: 10.1034/j.1600-0625.2002.110102.x
|
| [8] |
Seike M, Takata T, Ikeda M, et al. (2005) Histamine helps development of eczematous lesions in experimental contact dermatitis in mice. Arch Dermatol Res 297: 68–74. doi: 10.1007/s00403-005-0569-5
|
| [9] |
Seike M, Furuya K, Omura M, et al. (2010) Histamine H4 receptor antagonist ameliorates chronic allergic contact dermatitis induced by repeated challenge. Allergy 65: 319–326. doi: 10.1111/j.1398-9995.2009.02240.x
|
| [10] |
Matsushita A, Seike M, Okawa H, et al. (2012) Advantage of histamine H4 receptor antagonist usage with H1 receptor antagonist for the treatment of murine allergic contact dermatitis. Exp Dermatol 21: 714–715. doi: 10.1111/j.1600-0625.2012.01559.x
|
| [11] |
Ohsawa Y, Hirasawa N (2012) The antagonism of histamine H1 and H4 receptors ameliorates chronic allergic dermatitis via anti-pruritic and anti-inflammatory effects in NC/Nga mice. Allergy 67: 1014–1022. doi: 10.1111/j.1398-9995.2012.02854.x
|
| [12] |
Liang SC, Tan X, Luxenberg DP, et al. (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203: 2271–2279. doi: 10.1084/jem.20061308
|
| [13] |
Yi T, Chen Y, Wang L, et al. (2009) Reciprocal differentiation and tissue-specific pathogenesis of Th1, Th2 and Th17 cells in graft-versus-host disease. Blood 114: 3101–3112. doi: 10.1182/blood-2009-05-219402
|
| [14] |
Koga C, Kabashima K, Shiraishi N, et al. (2008) Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol 128: 2625–2630. doi: 10.1038/jid.2008.111
|
| [15] |
Matsushita A, Seike M, Hagiwara T, et al. (2014) Close relationship between T helper (Th)17 and Th2 response in murine allergic contact dermatitis. Clin Exp Dermatol 39: 924–931. doi: 10.1111/ced.12425
|
| [16] |
Ohtsu H, Tanaka S, Terui T, et al. (2001) Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS Lett 502: 53–56. doi: 10.1016/S0014-5793(01)02663-1
|
| [17] |
Tamura T, Matsubara M, Takada C (2004) Effects of olopatadine hydrochloride, antihistamine drug, on skin inflammation induced by repeated topical application of oxazolone in mice. Brit J Dermatol 151: 1133–1142. doi: 10.1111/j.1365-2133.2004.06172.x
|
| [18] |
Kim JY, Jeong MS, Park MK, et al. (2014) Time-dependent progression from the acute to chronic phases in atopic dermatitis induced by epicutaneous allergen stimulation in NC/Nga mice. Exp Dermatol 23: 53–57. doi: 10.1111/exd.12297
|
| [19] |
Ohtsu H, Kuramasu K, Tanaka S, et al. (2002) Plasma extravasation induced by dietary supplemented histamine in histamine-free mice. Eur J Immunol 32: 1698–1708. doi: 10.1002/1521-4141(200206)32:6<1698::AID-IMMU1698>3.0.CO;2-7
|
| [20] |
Hamada R, Seike M, Kamijima R, et al. (2006) Neuronal conditions of spinal cord in dermatitis are improved by olopatadine. Eur J Pharmacol 547: 45–51. doi: 10.1016/j.ejphar.2006.06.058
|
| [21] |
Tamaka K, Seike M, Hagiwara T, et al. (2015) Histamine suppresses regulatory T cells mediated by TGF-β in murine chronic contact dermatitis. Exp Dermatol 24: 280–284. doi: 10.1111/exd.12644
|
| [22] |
Nakae S, Komiyama Y, Nambu A, et al. (2002) Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17: 375–387. doi: 10.1016/S1074-7613(02)00391-6
|
| [23] |
Zhao Y, Balato A, Fishelevich R, et al. (2009) Th17/Tc17 infiltration and associated cytokine gene expression in elicitation phase of allergic contact dermatitis. Brit J Dermatol 161: 1301–1306. doi: 10.1111/j.1365-2133.2009.09400.x
|
| [24] | Kim D, McAlees JW, Bischoff LJ, et al. (2018) Combined administration of anti-IL-13 and anti-IL-17A at individually sub-therapeutic doses limits asthma-like symptoms in a mouse model of Th2/Th17 high asthma. Clin Exp Allergy, 24. |
| [25] |
Bian R, Tang J, Hu L, et al. (2018) (E)-phenethyl 3-(3,5-dihydroxy-4-isopropylphenyl) acrylate gel improves DNFB-induced allergic contact hypersensitivity via regulating the balance of Th1/Th2/T17/Treg cell subsets. Int Immunopharmacol 65: 8–15. doi: 10.1016/j.intimp.2018.09.032
|
| [26] |
Wu R, Zeng J, Yuan J, et al. (2018) MicroRNA-210 overexpression promotes psoriasis-like inflammation by inducing Th1 and Th17 cell differentiation. J Clin Invest 128: 2551–2568. doi: 10.1172/JCI97426
|
| [27] |
Orciani M, Campanati A, Caffarini M, et al. (2017) T helper (Th)1, Th17 and Th2 imbalance in mesenchymal stem cells of adult patients with atopic dermatitis: at the origin of the problem. Brit J Dermatol 176: 1569–1576. doi: 10.1111/bjd.15078
|
| [28] |
Harrington LE, Hatton RD, Mangan PR, et al. (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineage. Nat Immunol 6: 1123–1132. doi: 10.1038/ni1254
|
| [29] |
Mangan PR, Harrington LE, O'Quinn DB, et al. (2006) Transforming growth factor-beta induces development of the TH17 lineage. Nature 441: 231–234. doi: 10.1038/nature04754
|
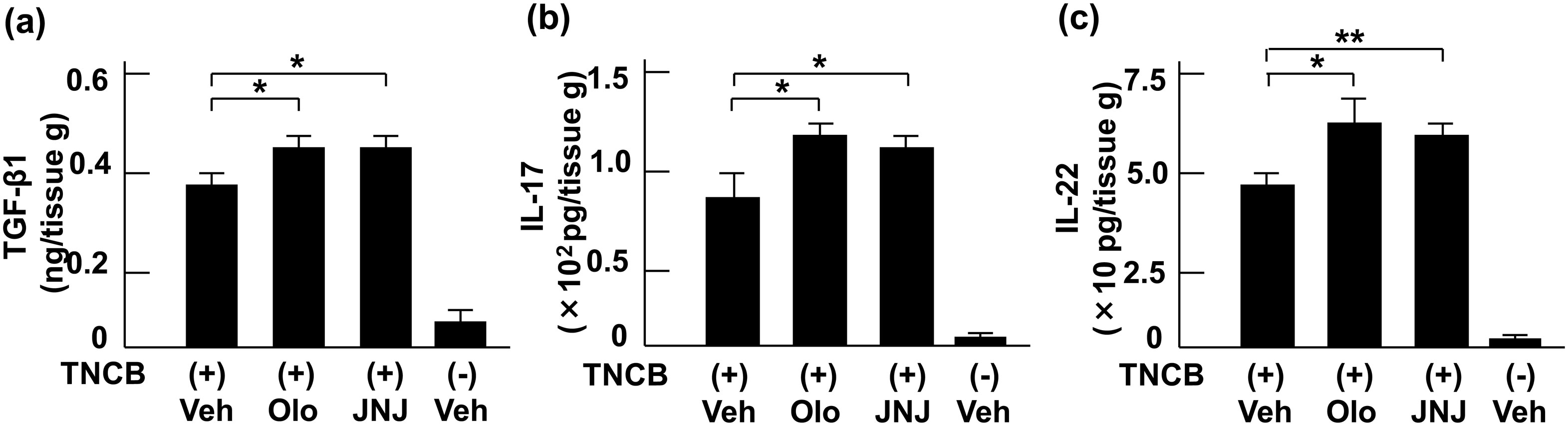
Figures(5)

Masahiro Seike. Histamine suppresses T helper 17 responses mediated by transforming growth factor-β1 in murine chronic allergic contact dermatitis[J]. AIMS Allergy and Immunology, 2018, 2(4): 180-189. doi: 10.3934/Allergy.2018.4.180







 DownLoad:
DownLoad: