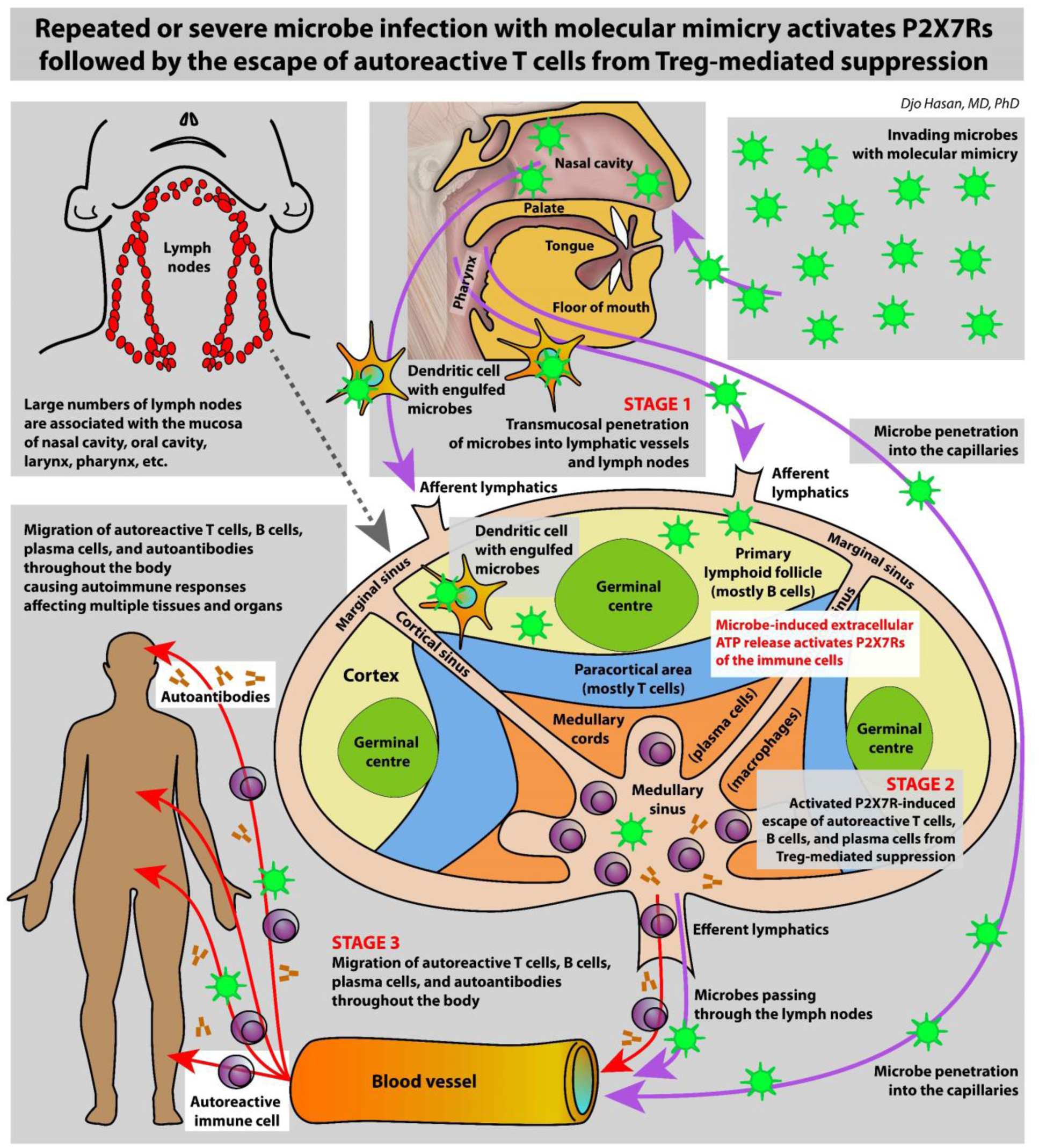
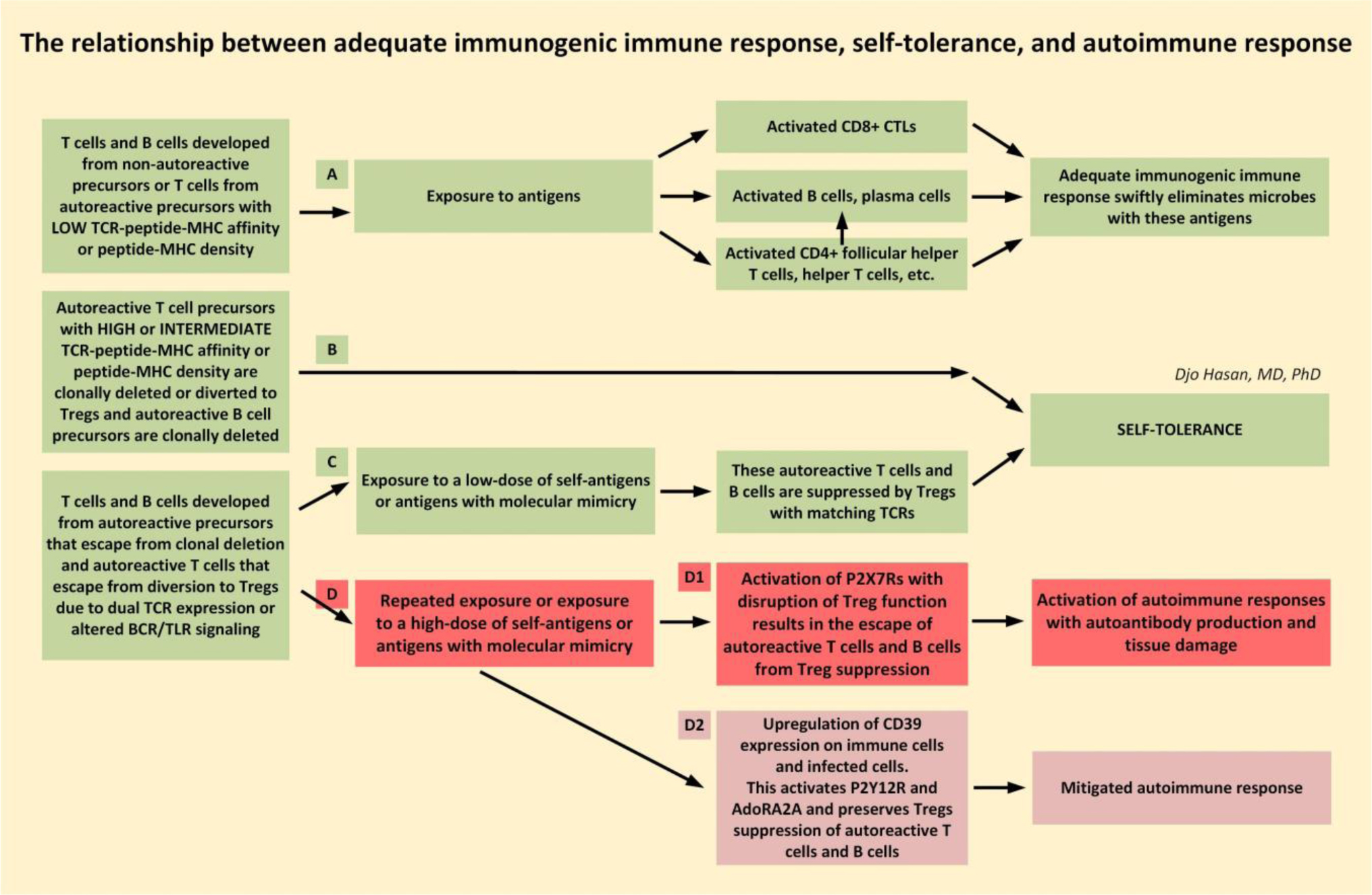
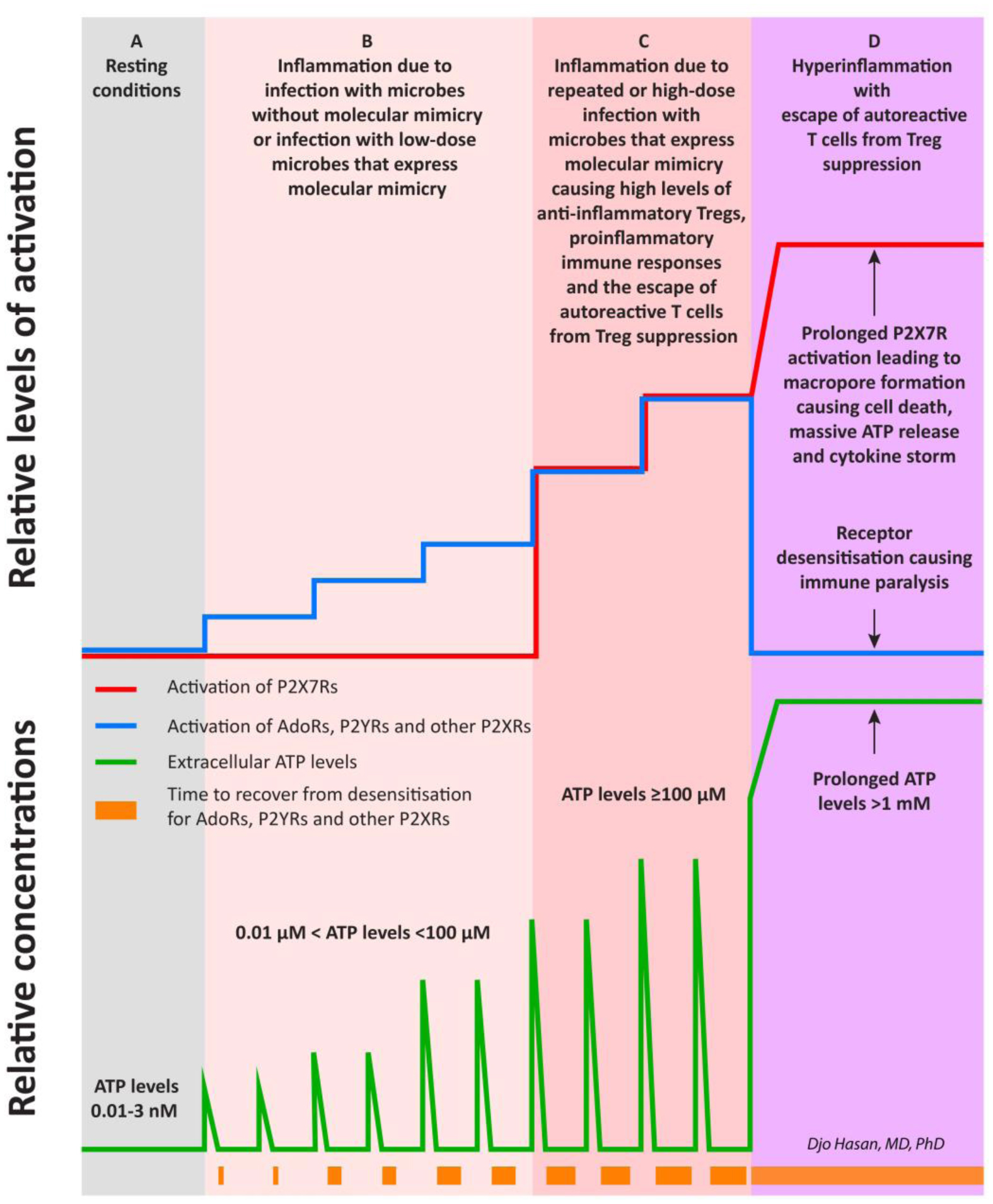
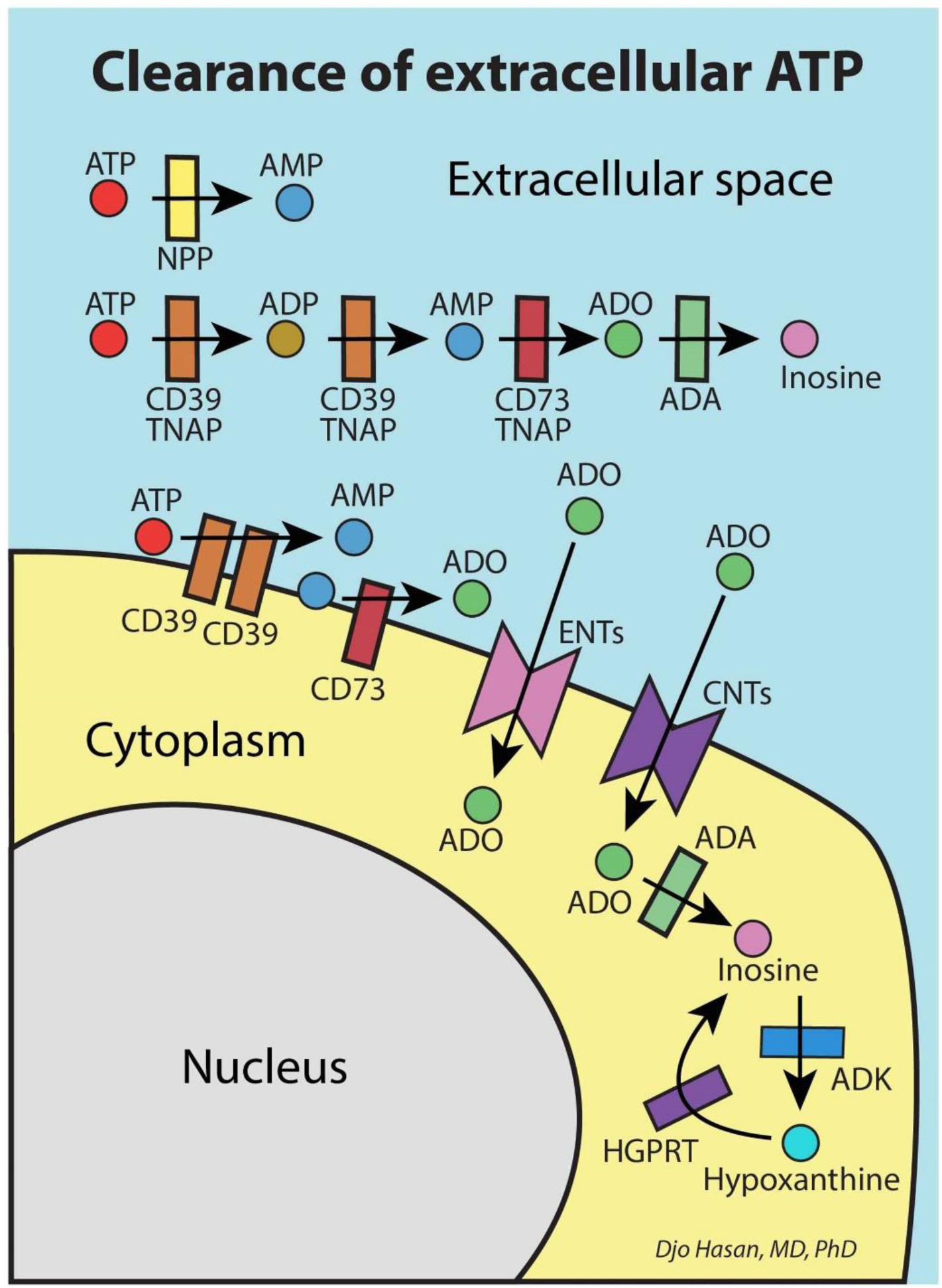
The immune system is meant to protect against invading microbes. Although this system is effective against many microbes, some can use molecular mimicry to turn the immune system against the host and activate autoimmune responses. The resulting autoimmunity has significant implications for public health and healthcare costs. It is well known that regulatory T cells (Tregs) are crucial for self-tolerance and that their function is impaired after exposure to self-antigens or antigens with molecular mimicry, leading to the activation of autoimmune responses. How molecular mimicry disrupts Tregs in this manner remains under debate. This review contributes to the field of the pathogenesis of autoimmunity by proposing that purinergic signaling in the lymph nodes, with extracellular ATP, ADP, and adenosine as ligands, plays a pivotal role in this process. Repeated or high-dose microbial infection causes the release of large amounts of extracellular ATP sufficient to reach the threshold of extracellular ATP levels for activating P2X7 purinergic receptors (P2X7Rs) on dendritic cells and Tregs. This hampers the ability of Tregs to suppress autoimmune responses. Crucially, P2X7Rs are activated at very high extracellular ATP levels, thus only after repeated or high-dose infection with microbes. Arguably, in contrast to the rapid elimination of microbes with foreign antigens, the clearance of invading microbes that employ molecular mimicry requires the activation of P2X7Rs at the expense of self-tolerance. Because all processes required to activate autoimmune responses occur in secondary lymphoid organs, this article hypothesizes that, contrary to current convention, microbes do not need to enter organs to initiate autoimmunity. However, some types of microbes can prevent P2X7R-induced Treg disruption by converting extracellular ATP to adenosine, mitigating autoimmune responses resulting in chronic diseases with less severe inflammation. The proposed hypothesized mechanism has potential implications for the understanding and treatment of autoimmune disorders.
Citation: Djo Hasan. Purinergic P2X7R expressed on regulatory T cells potentially links molecular mimicry to autoimmune responses[J]. AIMS Allergy and Immunology, 2024, 8(2): 80-123. doi: 10.3934/Allergy.2024006
The immune system is meant to protect against invading microbes. Although this system is effective against many microbes, some can use molecular mimicry to turn the immune system against the host and activate autoimmune responses. The resulting autoimmunity has significant implications for public health and healthcare costs. It is well known that regulatory T cells (Tregs) are crucial for self-tolerance and that their function is impaired after exposure to self-antigens or antigens with molecular mimicry, leading to the activation of autoimmune responses. How molecular mimicry disrupts Tregs in this manner remains under debate. This review contributes to the field of the pathogenesis of autoimmunity by proposing that purinergic signaling in the lymph nodes, with extracellular ATP, ADP, and adenosine as ligands, plays a pivotal role in this process. Repeated or high-dose microbial infection causes the release of large amounts of extracellular ATP sufficient to reach the threshold of extracellular ATP levels for activating P2X7 purinergic receptors (P2X7Rs) on dendritic cells and Tregs. This hampers the ability of Tregs to suppress autoimmune responses. Crucially, P2X7Rs are activated at very high extracellular ATP levels, thus only after repeated or high-dose infection with microbes. Arguably, in contrast to the rapid elimination of microbes with foreign antigens, the clearance of invading microbes that employ molecular mimicry requires the activation of P2X7Rs at the expense of self-tolerance. Because all processes required to activate autoimmune responses occur in secondary lymphoid organs, this article hypothesizes that, contrary to current convention, microbes do not need to enter organs to initiate autoimmunity. However, some types of microbes can prevent P2X7R-induced Treg disruption by converting extracellular ATP to adenosine, mitigating autoimmune responses resulting in chronic diseases with less severe inflammation. The proposed hypothesized mechanism has potential implications for the understanding and treatment of autoimmune disorders.

| [1] |
Miller FW (2023) The increasing prevalence of autoimmunity and autoimmune diseases: an urgent call to action for improved understanding, diagnosis, treatment, and prevention. Curr Opin Immunol 80: 102266. https://doi.org/10.1016/j.coi.2022.102266 
|
| [2] | Goldhaft TM, Wernicoff N (1948) High mortality associated with a widespread outbreak of Newcastle disease. Cornell Vet 38: 181-185. |
| [3] |
Nelson CB, Pomeroy BS, Schrall K, et al. (1952) An outbreak of conjunctivitis due to Newcastle disease virus (NDV) occurring in poultry workers. Am J Public Health Nations Health 42: 672-678. https://doi.org/10.2105/AJPH.42.6.672
|
| [4] |
Trott DG, Pilsworth R (1965) Outbreaks of conjunctivitis due to the Newcastle disease virus among workers in chicken-broiler factories. Br Med J 2: 1514-1517. https://doi.org/10.1136/bmj.2.5477.1514
|
| [5] |
Rojas M, Restrepo-Jiménez P, Monsalve DM, et al. (2018) Molecular mimicry and autoimmunity. J Autoimmun 95: 100-123. https://doi.org/10.1016/j.jaut.2018.10.012
|
| [6] |
Suliman BA (2024) Potential clinical implications of molecular mimicry-induced autoimmunity. Immun Inflamm Dis 12: e1178. https://doi.org/10.1002/iid3.1178
|
| [7] |
Kanduc D, Shoenfeld Y (2020) Medical, genomic, and evolutionary aspects of the peptide Sharing between Pathogens, Primates, and Humans. Glob Med Genet 7: 64-67. https://doi.org/10.1055/s-0040-1716334
|
| [8] |
Oldstone MB (2014) Molecular mimicry: its evolution from concept to mechanism as a cause of autoimmune diseases. Monoclon Antib Immunodiagn Immunother 33: 158-165. https://doi.org/10.1089/mab.2013.0090
|
| [9] |
Santambrogio L, Marrack P (2023) The broad spectrum of pathogenic autoreactivity. Nat Rev Immunol 23: 69-70. https://doi.org/10.1038/s41577-022-00812-2
|
| [10] |
Simon CE (1909) On auto-antibody formation and antihemolysis. J Exp Med 11: 695-717. https://doi.org/10.1084/jem.11.5.695
|
| [11] |
Schwentker FF, Rivers TM (1934) The antibody response of rabbits to injections of emulsions and extracts of homologous brain. J Exp Med 60: 559-574. https://doi.org/10.1084/jem.60.5.559
|
| [12] |
Gear J (1945) Autoantigens and autoantibodies in the pathogenesis of disease with special refenence to blackwater fever. Trans R Soc Trop Med Hyg 39: 301-314. https://doi.org/10.1016/0035-9203(46)90041-7
|
| [13] |
Cavelti PA (1945) Autoantibodies in rheumatic fever. Proc Soc Exp Biol Med 60: 379-381. https://doi.org/10.3181/00379727-60-15197P
|
| [14] |
Liblau RS, Wong FS, Mars LT, et al. (2002) Autoreactive CD8 T cells in organ-specific autoimmunity: Emerging targets for therapeutic intervention. Immunity 17: 1-6. https://doi.org/10.1016/S1074-7613(02)00338-2
|
| [15] |
Shepard ER, Wegner A, Hill EV, et al. (2021) The mechanism of action of antigen processing independent T cell epitopes designed for immunotherapy of autoimmune diseases. Front Immunol 12: 654201. https://doi.org/10.3389/fimmu.2021.654201
|
| [16] |
Klein L, Robey EA, Hsieh CS (2019) Central CD4(+) T cell tolerance: deletion versus regulatory T cell differentiation. Nat Rev Immunol 19: 7-18. https://doi.org/10.1038/s41577-018-0083-6
|
| [17] |
Sharma C, Bayry J (2023) High risk of autoimmune diseases after COVID-19. Nat Rev Rheumatol 19: 399-400. https://doi.org/10.1038/s41584-023-00964-y
|
| [18] |
Lerner A, Benzvi C, Vojdani A (2023) Cross-reactivity and sequence similarity between microbial transglutaminase and human tissue antigens. Sci Rep 13: 17526. https://doi.org/10.1038/s41598-023-44452-5
|
| [19] | Trier NH, Houen G (2023) Antibody cross-reactivity in auto-immune diseases. Int J Mol Sci 24. https://doi.org/10.3390/ijms241713609 |
| [20] |
English J, Patrick S, Stewart LD (2023) The potential role of molecular mimicry by the anaerobic microbiota in the aetiology of autoimmune disease. Anaerobe 80: 102721. https://doi.org/10.1016/j.anaerobe.2023.102721
|
| [21] |
Pisetsky DS (2023) Pathogenesis of autoimmune disease. Nat Rev Nephrol 19: 509-524. https://doi.org/10.1038/s41581-023-00720-1
|
| [22] |
Hasan D, Shono A, van Kalken CK, et al. (2022) A novel definition and treatment of hyperinflammation in COVID-19 based on purinergic signalling. Purinergic Signal 18: 13-59. https://doi.org/10.1007/s11302-021-09814-6
|
| [23] |
Jerne NK (1955) The natural-selection theory of antibody formation. Proc Natl Acad Sci USA 41: 849-857. https://doi.org/10.1073/pnas.41.11.849
|
| [24] |
Burnet FM (1976) A modification of Jerne's theory of antibody production using the concept of clonal selection. CA Cancer J Clin 26: 119-121. https://doi.org/10.3322/canjclin.26.2.119
|
| [25] | Burnet FM (1957) A modification of Jerne's theory of antibody production using the concept of clonal selection. Aust J Sci 20: 67-69. |
| [26] |
Rajendeeran A, Tenbrock K (2021) Regulatory T cell function in autoimmune disease. J Transl Autoimmun 4: 100130. https://doi.org/10.1016/j.jtauto.2021.100130
|
| [27] |
Dominguez-Villar M, Hafler DA (2018) Regulatory T cells in autoimmune disease. Nat Immunol 19: 665-673. https://doi.org/10.1038/s41590-018-0120-4
|
| [28] |
Santori FR, Arsov I, Lilić M, et al. (2002) Editing autoreactive TCR enables efficient positive selection. J Immunol 169: 1729-1734. https://doi.org/10.4049/jimmunol.169.4.1729
|
| [29] |
Zal T, Weiss S, Mellor A, et al. (1996) Expression of a second receptor rescues self-specific T cells from thymic deletion and allows activation of autoreactive effector function. Proc Natl Acad Sci USA 93: 9102-9107. https://doi.org/10.1073/pnas.93.17.9102
|
| [30] |
Ni PP, Solomon B, Hsieh CS, et al. (2014) The ability to rearrange dual TCRs enhances positive selection, leading to increased Allo- and Autoreactive T cell repertoires. J Immunol 193: 1778-1786. https://doi.org/10.4049/jimmunol.1400532
|
| [31] |
Auger JL, Haasken S, Steinert EM, et al. (2012) Incomplete TCR-β allelic exclusion accelerates spontaneous autoimmune arthritis in K/BxN TCR transgenic mice. Eur J Immunol 42: 2354-2362. https://doi.org/10.1002/eji.201242520
|
| [32] |
George TC, Bilsborough J, Viney JL, et al. (2003) High antigen dose and activated dendritic cells enable Th cells to escape regulatory T cell-mediated suppression in vitro. Eur J Immunol 33: 502-511. https://doi.org/10.1002/immu.200310026
|
| [33] |
Swee LK, Nusser A, Curti M, et al. (2014) The amount of self-antigen determines the effector function of murine T cells escaping negative selection. Eur J Immunol 44: 1299-1312.
|
| [34] |
Davis MM (2015) Not-so-negative selection. Immunity 43: 833-835. https://doi.org/10.1002/eji.201343840
|
| [35] |
Legoux FP, Lim JB, Cauley AW, et al. (2015) CD4+ T cell tolerance to tissue-restricted self antigens is mediated by antigen-specific regulatory T cells rather than deletion. Immunity 43: 896-908. https://doi.org/10.1016/j.immuni.2015.10.011
|
| [36] |
Rawlings DJ, Metzler G, Wray-Dutra M, et al. (2017) Altered B cell signalling in autoimmunity. Nat Rev Immunol 17: 421-436. https://doi.org/10.3390/cells10051190
|
| [37] |
Bonasia CG, Abdulahad WH, Rutgers A, et al. (2021) B cell activation and escape of tolerance checkpoints: Recent insights from studying autoreactive B cells. Cells 10: 1190. https://doi.org/10.3390/cells10051190
|
| [38] |
Giltiay NV, Chappell CP, Clark EA (2012) B-cell selection and the development of autoantibodies. Arthritis Res Ther 14. https://doi.org/10.1186/ar3918
|
| [39] |
Suurmond J, Diamond B (2015) Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest 125: 2194-2202. https://doi.org/10.1172/JCI78084
|
| [40] |
Sakaguchi S, Wing K, Miyara M (2007) Regulatory T cells–a brief history and perspective. Eur J Immunol 37: S116-S123. https://doi.org/10.1002/eji.200737593
|
| [41] |
Wing JB, Tekgüç M, Sakaguchi S (2018) Control of germinal center responses by T-Follicular regulatory cells. Front Immunol 9: 1910. https://doi.org/10.3389/fimmu.2018.01910
|
| [42] |
Wing JB, Lim EL, Sakaguchi S (2020) Control of foreign Ag-specific Ab responses by Treg and Tfr. Immunol Rev 296: 104-119. https://doi.org/10.1111/imr.12888
|
| [43] |
Ding T, Su R, Wu R, et al. (2021) Frontiers of autoantibodies in autoimmune disorders: Crosstalk between Tfh/Tfr and regulatory B cells. Front Immunol 12: 641013. https://doi.org/10.3389/fimmu.2021.641013
|
| [44] |
Richards DM, Delacher M, Goldfarb Y, et al. (2015) Chapter eight-Treg cell differentiation: from thymus to peripheral tissue. Progress in Molecular Biology and Translational Science . New York: Academic Press 175-205. https://doi.org/10.1016/bs.pmbts.2015.07.014
|
| [45] |
Schmitt EG, Williams CB (2013) Generation and function of induced regulatory T cells. Front Immunol 4: 152. https://doi.org/10.3389/fimmu.2013.00152
|
| [46] |
Khantakova JN, Bulygin AS, Sennikov SV (2022) The regulatory-T-cell memory phenotype: What we know. Cells 11. https://doi.org/10.3390/cells11101687
|
| [47] |
Sanchez AM, Zhu J, Huang X, et al. (2012) The development and function of memory regulatory T cells after acute viral infections. J Immunol 189: 2805-2814. https://doi.org/10.4049/jimmunol.1200645
|
| [48] |
Rosenblum MD, Way SS, Abbas AK (2016) Regulatory T cell memory. Nat Rev Immunol 16: 90-101. https://doi.org/10.1038/nri.2015.1
|
| [49] |
Kaminski A, Hager FT, Kopplin L, et al. (2023) Resident regulatory T cells reflect the immune history of individual lymph nodes. Sci Immunol 8: eadj5789. https://doi.org/10.1126/sciimmunol.adj5789
|
| [50] |
Corthay A (2009) How do regulatory T cells work?. Scand J Immunol 70: 326-336. https://doi.org/10.1111/j.1365-3083.2009.02308.x
|
| [51] |
Sprouse ML, Scavuzzo MA, Blum S, et al. (2018) High self-reactivity drives T-bet and potentiates Treg function in tissue-specific autoimmunity. JCI Insight 3. https://doi.org/10.1172/jci.insight.97322
|
| [52] |
Zuany-Amorim C, Sawicka E, Manlius C, et al. (2002) Suppression of airway eosinophilia by killed Mycobacterium vaccae-induced allergen-specific regulatory T-cells. Nat Med 8: 625-629. https://doi.org/10.1038/nm0602-625
|
| [53] |
Belkaid Y, Piccirillo CA, Mendez S, et al. (2002) CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420: 502-507. https://doi.org/10.1038/nature01152
|
| [54] |
Suffia IJ, Reckling SK, Piccirillo CA, et al. (2006) Infected site-restricted Foxp3+ natural regulatory T cells are specific for microbial antigens. J Exp Med 203: 777-788. https://doi.org/10.1084/jem.20052056
|
| [55] |
Montagnoli C, Bacci A, Bozza S, et al. (2002) B7/CD28-dependent CD4+CD25+ regulatory T cells are essential components of the memory-protective immunity to Candida albicans. J Immunol 169: 6298-6308. https://doi.org/10.4049/jimmunol.169.11.6298
|
| [56] |
McGuirk P, McCann C, Mills KH (2002) Pathogen-specific T regulatory 1 cells induced in the respiratory tract by a bacterial molecule that stimulates interleukin 10 production by dendritic cells: a novel strategy for evasion of protective T helper type 1 responses by Bordetella pertussis. J Exp Med 195: 221-231. https://doi.org/10.1084/jem.20011288
|
| [57] |
Kullberg MC, Jankovic D, Gorelick PL, et al. (2002) Bacteria-triggered CD4(+) T regulatory cells suppress Helicobacter hepaticus-induced colitis. J Exp Med 196: 505-515. https://doi.org/10.1084/jem.20020556
|
| [58] |
Ochando JC, Yopp AC, Yang Y, et al. (2005) Lymph node occupancy is required for the peripheral development of alloantigen-specific Foxp3+ regulatory T cells. J Immunol 174: 6993-7005. https://doi.org/10.4049/jimmunol.174.11.6993
|
| [59] |
van Maurik A, Herber M, Wood KJ, et al. (2002) Cutting edge: CD4+CD25+ alloantigen-specific immunoregulatory cells that can prevent CD8+ T cell-mediated graft rejection: implications for anti-CD154 immunotherapy. J Immunol 169: 5401-5404. https://doi.org/10.4049/jimmunol.169.10.5401
|
| [60] |
Smith DG, Martinelli R, Besra GS, et al. (2019) Identification and characterization of a novel anti-inflammatory lipid isolated from Mycobacterium vaccae, a soil-derived bacterium with immunoregulatory and stress resilience properties. Psychopharmacology 236: 1653-1670. https://doi.org/10.1007/s00213-019-05253-9
|
| [61] |
Múnera M, Farak J, Pérez M, et al. (2020) Prediction of molecular mimicry between antigens from Leishmania sp. and human: Implications for autoimmune response in systemic lupus erythematosus. Microb Pathog 148: 104444. https://doi.org/10.1016/j.micpath.2020.104444
|
| [62] | Argov S, Jaffe CL, Krupp M, et al. (1989) Autoantibody production by patients infected with Leishmania. Clin Exp Immunol 76: 190-197. |
| [63] |
Gustafson KS, Vercellotti GM, Bendel CM, et al. (1991) Molecular mimicry in Candida albicans. Role of an integrin analogue in adhesion of the yeast to human endothelium. J Clin Invest 87: 1896-1902. https://doi.org/10.1172/JCI115214
|
| [64] | Vojdani A, Rahimian P, Kalhor H, et al. (1996) Immunological cross reactivity between Candida albicans and human tissue. J Clin Lab Immunol 48: 1-15. |
| [65] |
Sandros J, Rozdzinski E, Zheng J, et al. (1994) Lectin domains in the toxin of Bordetella pertussis: selectin mimicry linked to microbial pathogenesis. Glycoconj J 11: 501-506. https://doi.org/10.1007/BF00731300
|
| [66] |
Sandros J, Tuomanen E (1993) Attachment factors of Bordetella pertussis: mimicry of eukaryotic cell recognition molecules. Trends Microbiol 1: 192-196. https://doi.org/10.1016/0966-842X(93)90090-E
|
| [67] |
Alyamani EJ, Brandt P, Pena JA, et al. (2007) Helicobacter hepaticus catalase shares surface-predicted epitopes with mammalian catalases. Microbiology (Reading) 153: 1006-1016. https://doi.org/10.1099/mic.0.29184-0
|
| [68] |
Macdonald WA, Chen Z, Gras S, et al. (2009) T cell allorecognition via molecular mimicry. Immunity 31: 897-908. https://doi.org/10.1016/j.immuni.2009.09.025
|
| [69] |
Koyama M, Hill GR (2016) Alloantigen presentation and graft-versus-host disease: fuel for the fire. Blood 127: 2963-2970. https://doi.org/10.1182/blood-2016-02-697250
|
| [70] |
Hudson KE, Wong ASL, Richards AL, et al. (2019) Alloimmunogenicity of an isolated MHC allele is affected by the context of MHC mismatch in a murine model. Transfusion 59: 744-753. https://doi.org/10.1111/trf.15109
|
| [71] |
Mondino S, Schmidt S, Buchrieser C (2020) Molecular mimicry: A paradigm of host-microbe coevolution illustrated by legionella. mBio 11. https://doi.org/10.1128/mBio.01201-20
|
| [72] |
Pacholczyk R, Kern J, Singh N, et al. (2007) Nonself-antigens are the cognate specificities of Foxp3+ regulatory T cells. Immunity 27: 493-504. https://doi.org/10.1016/j.immuni.2007.07.019
|
| [73] |
Stephens GL, Shevach EM (2007) Foxp3+ regulatory T cells: selfishness under scrutiny. Immunity 27: 417-419.
|
| [74] | Vignali DA, Collison LW, Workman CJ (2008) How regulatory T cells work. NatRev Immunol 8: 523-532. https://doi.org/10.1038/nri2343 |
| [75] |
Tan D, Yin W, Guan F, et al. (2022) B cell-T cell interplay in immune regulation: A focus on follicular regulatory T and regulatory B cell functions. Front Cell Dev Biol 10: 991840. https://doi.org/10.3389/fcell.2022.991840
|
| [76] |
Rosser EC, Mauri C (2015) Regulatory B cells: origin, phenotype, and function. Immunity 42: 607-612. https://doi.org/10.1016/j.immuni.2015.04.005
|
| [77] |
Wang R, Xie R, Song Z (2018) Circulating regulatory Tfh cells are enriched in patients with chronic hepatitis B infection and induce the differentiation of regulatory B cells. Exp Cell Res 365: 171-176. https://doi.org/10.1016/j.yexcr.2018.02.031
|
| [78] |
Li H, Zhou R, Wang C, et al. (2018) T follicular regulatory cells infiltrate the human airways during the onset of acute respiratory distress syndrome and regulate the development of B regulatory cells. Immunol Res 66: 548-554. https://doi.org/10.1007/s12026-018-9014-7
|
| [79] |
Catalán D, Mansilla MA, Ferrier A, et al. (2021) Immunosuppressive mechanisms of regulatory B Cells. Front Immunol 12. https://doi.org/10.3389/fimmu.2021.611795
|
| [80] |
Figueiró F, Muller L, Funk S, et al. (2016) Phenotypic and functional characteristics of CD39 (high) human regulatory B cells (Breg). Oncoimmunology 5: e1082703. https://doi.org/10.1080/2162402X.2015.1082703
|
| [81] |
Kaku H, Cheng KF, Al-Abed Y, et al. (2014) A novel mechanism of B cell-mediated immune suppression through CD73 expression and adenosine production. J Immunol 193: 5904-5913. https://doi.org/10.4049/jimmunol.1400336
|
| [82] |
Saze Z, Schuler PJ, Hong CS, et al. (2013) Adenosine production by human B cells and B cell-mediated suppression of activated T cells. Blood 122: 9-18. https://doi.org/10.1182/blood-2013-02-482406
|
| [83] |
Khan AR, Hams E, Floudas A, et al. (2015) PD-L1hi B cells are critical regulators of humoral immunity. Nat Commun 6: 5997. https://doi.org/10.1038/ncomms6997
|
| [84] |
Wang K, Tao L, Su J, et al. (2017) TLR4 supports the expansion of FasL(+)CD5(+)CD1d(hi) regulatory B cells, which decreases in contact hypersensitivity. Mol Immunol 87: 188-199. https://doi.org/10.1016/j.molimm.2017.04.016
|
| [85] |
Ma L, Liu B, Jiang Z, et al. (2014) Reduced numbers of regulatory B cells are negatively correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin Rheumatol 33: 187-195. https://doi.org/10.1007/s10067-013-2359-3
|
| [86] |
Yang M, Deng J, Liu Y, et al. (2012) IL-10-producing regulatory B10 cells ameliorate collagen-induced arthritis via suppressing Th17 cell generation. Am J Pathol 180: 2375-2385. https://doi.org/10.1016/j.ajpath.2012.03.010
|
| [87] |
Flores-Borja F, Bosma A, Ng D, et al. (2013) CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med 5: 173ra123. https://doi.org/10.1126/scitranslmed.3005407
|
| [88] |
Carter NA, Rosser EC, Mauri C (2012) Interleukin-10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis Res Ther 14: R32. https://doi.org/10.1186/ar3736
|
| [89] |
Gondek DC, Lu LF, Quezada SA, et al. (2005) Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol 174: 1783-1786. https://doi.org/10.4049/jimmunol.174.4.1783
|
| [90] |
Jahrsdörfer B, Vollmer A, Blackwell SE, et al. (2010) Granzyme B produced by human plasmacytoid dendritic cells suppresses T-cell expansion. Blood 115: 1156-1165. https://doi.org/10.1182/blood-2009-07-235382
|
| [91] |
Xu L, Liu X, Liu H, et al. (2017) Impairment of granzyme B-producing regulatory B cells correlates with exacerbated rheumatoid arthritis. Front Immunol 8: 768. https://doi.org/10.3389/fimmu.2017.00768
|
| [92] | Saraiva M, Vieira P, O'Garra A (2019) Biology and therapeutic potential of interleukin-10. J Exp Med 217. https://doi.org/10.1084/jem.20190418 |
| [93] |
Zysk W, Gleń J, Trzeciak M (2022) Current insight into the role of IL-35 and its potential involvement in the pathogenesis and therapy of atopic dermatitis. Int J Mol Sci 23: 15709. https://doi.org/10.3390/ijms232415709
|
| [94] |
Massagué J, Sheppard D (2023) TGF-β signaling in health and disease. Cell 186: 4007-4037. https://doi.org/10.1016/j.cell.2023.07.036
|
| [95] |
Needham EJ, Stoevesandt O, Thelin EP, et al. (2021) Complex autoantibody responses occur following moderate to severe traumatic brain injury. J Immunol 207: 90-100. https://doi.org/10.4049/jimmunol.2001309
|
| [96] |
Zhang Z, Zoltewicz JS, Mondello S, et al. (2014) Human traumatic brain injury induces autoantibody response against glial fibrillary acidic protein and its breakdown products. PLoS One 9: e92698. https://doi.org/10.1371/journal.pone.0092698
|
| [97] | Zimering MB (2023) Repeated traumatic brain injury is associated with neurotoxic plasma autoantibodies directed against the serotonin 2A and alpha 1 adrenergic receptors. Endocrinol Diabetes Metab J 7: 1-12. https://doi.org/10.31038/EDMJ.2023722 |
| [98] |
Zimering MB, Pulikeyil AT, Myers CE, et al. (2020) Serotonin 2A receptor autoantibodies increase in adult traumatic brain injury in association with neurodegeneration. J Endocrinol Diabetes 7: 1-8. https://doi.org/10.15226/2374-6890/7/1/001142
|
| [99] |
Vijapur SM, Yang Z, Barton DJ, et al. (2020) Anti-pituitary and anti-hypothalamus autoantibody associations with inflammation and persistent hypogonadotropic hypogonadism in men with traumatic brain injury. J Neurotrauma 37: 1609-1626. https://doi.org/10.1089/neu.2019.6780
|
| [100] |
Arevalo-Martin A, Grassner L, Garcia-Ovejero D, et al. (2018) Elevated autoantibodies in subacute human spinal cord injury are naturally occurring antibodies. Front Immunol 9. https://doi.org/10.3389/fimmu.2018.02365
|
| [101] |
Schwab JM, Haider C, Kopp MA, et al. (2023) Lesional antibody synthesis and complement deposition associate with de novo antineuronal antibody synthesis after spinal cord injury. Neurol Neuroimmunol Neuroinflammation 10: e200099. https://doi.org/10.1212/NXI.0000000000200099
|
| [102] |
Caputo F, Barranco R, Ricci P, et al. (2021) An unusual case of coma related to glutamate receptor 3 (GluR3) auto-antibodies after a traumatic spinal cord injury: Clinical and pathological aspects. Med Leg J 89: 133-136. https://doi.org/10.1177/0025817220970069
|
| [103] |
Davies AL, Hayes KC, Dekaban GA (2007) Clinical correlates of elevated serum concentrations of cytokines and autoantibodies in patients with spinal cord injury. Arch Phys Med Rehabil 88: 1384-1393. https://doi.org/10.1016/j.apmr.2007.08.004
|
| [104] |
Hayes KC, Hull TC, Delaney GA, et al. (2002) Elevated serum titers of proinflammatory cytokines and CNS autoantibodies in patients with chronic spinal cord injury. J Neurotrauma 19: 753-761. https://doi.org/10.1089/08977150260139129
|
| [105] |
Panoutsakopoulou V, Sanchirico ME, Huster KM, et al. (2001) Analysis of the relationship between viral infection and autoimmune disease. Immunity 15: 137-147. https://doi.org/10.1016/S1074-7613(01)00172-8
|
| [106] |
Wang EY, Mao T, Klein J, et al. (2021) Diverse functional autoantibodies in patients with COVID-19. Nature 595: 283-288. https://doi.org/10.1038/s41586-021-03631-y
|
| [107] |
Trahtemberg U, Rottapel R, Dos Santos CC, et al. (2021) Anticardiolipin and other antiphospholipid antibodies in critically ill COVID-19 positive and negative patients. Ann Rheum Dis 80: 1236-1240. https://doi.org/10.1136/annrheumdis-2021-220206
|
| [108] |
Chang SE, Feng A, Meng W, et al. (2021) New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat Commun 12: 5417. https://doi.org/10.1038/s41467-021-25509-3
|
| [109] |
Arthur JM, Forrest JC, Boehme KW, et al. (2021) Development of ACE2 autoantibodies after SARS-CoV-2 infection. PLoS One 16: e0257016. https://doi.org/10.1371/journal.pone.0257016
|
| [110] |
Humphrey JH (1948) The pathogenesis of glomerulonephritis; a reinvestigation of the auto-immunization hypothesis. J Pathol Bacteriol 60: 211-218. https://doi.org/10.1002/path.1700600208
|
| [111] | Cavelti PA (1947) Studies on the pathogenesis of rheumatic fever; experimental production of autoantibodies to heart, skeletal muscle and connective tissue. Arch Pathol (Chic) 44: 1-12. |
| [112] | Cavelti PA (1947) Studies on the pathogenesis of rheumatic fever; cardiac lesions produced in rats by means of autoantibodies to heart and connective tissues. Arch Pathol (Chic) 44: 13-27. |
| [113] | Cavelti PA (1947) Pathogenesis of glomerulonephritis and rheumatic fever; in vivo activation of tissue antigens as a result of streptococcic infection and consecutive formation of autoantibodies. Arch Pathol (Chic) 44: 119-125. |
| [114] |
Segal Y, Shoenfeld Y (2018) Vaccine-induced autoimmunity: the role of molecular mimicry and immune crossreaction. Cell Mol Immunol 15: 586-594. https://doi.org/10.1038/cmi.2017.151
|
| [115] |
Talotta R (2023) Molecular mimicry and HLA polymorphisms may drive autoimmunity in recipients of the BNT-162b2 mRNA vaccine: A computational analysis. Microorganisms 11. https://doi.org/10.3390/microorganisms11071686
|
| [116] |
Franco A, Song J, Chambers C, et al. (2023) SARS-CoV-2 spike-specific regulatory T cells (Treg) expand and develop memory in vaccine recipients suggesting a role for immune regulation in preventing severe symptoms in COVID-19. Autoimmunity 56: 2259133. https://doi.org/10.1080/08916934.2023.2259133
|
| [117] |
Fiorelli D, Caruso V, Belardi R, et al. (2023) Evaluation of autoantibody profile in healthy subjects after mRNA vaccination against COVID-19. Int Immunopharmacol 122: 110592. https://doi.org/10.1016/j.intimp.2023.110592
|
| [118] |
Greinacher A, Selleng K, Palankar R, et al. (2021) Insights in ChAdOx1 nCoV-19 vaccine-induced immune thrombotic thrombocytopenia. Blood 138: 2256-2268. https://doi.org/10.1182/blood.2021013231
|
| [119] |
Greinacher A, Thiele T, Warkentin TE, et al. (2021) Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N Engl J Med 384: 2092-2101. https://doi.org/10.1056/NEJMoa2104840
|
| [120] |
Krashias G, Pafiti A, Deeba E, et al. (2022) SARS CoV-2 vaccination induces antibodies against cardiolipin. BMC Res Notes 15: 292. https://doi.org/10.1186/s13104-022-06180-3
|
| [121] |
Xu W, Wen X, Cong X, et al. (2023) COVID-19 mRNA vaccine, but not a viral vector-based vaccine, promotes neutralizing anti-type I interferon autoantibody production in a small group of healthy individuals. J Med Virol 95: e29137. https://doi.org/10.1002/jmv.29137
|
| [122] |
Aladdin Y, Algahtani H, Shirah B (2021) Vaccine-induced immune thrombotic thrombocytopenia with disseminated intravascular coagulation and death following the ChAdOx1 nCoV-19 vaccine. J Stroke Cerebrovasc Dis 30: 105938. https://doi.org/10.1016/j.jstrokecerebrovasdis.2021.105938
|
| [123] |
Alami A, Villeneuve PJ, Farrell PJ, et al. (2023) Myocarditis and pericarditis post-mRNA COVID-19 vaccination: Insights from a pharmacovigilance perspective. J Clin Med 12. https://doi.org/10.3390/jcm12154971
|
| [124] | Di Dedda EA, Barison A, Aquaro GD, et al. (2022) Cardiac magnetic resonance imaging of myocarditis and pericarditis following COVID-19 vaccination: a multicenter collection of 27 cases. Eur Radiol : 1-9. https://doi.org/10.1007/s00330-022-08566-0 |
| [125] |
Yap J, Tham MY, Poh J, et al. (2022) Pericarditis and myocarditis after COVID-19 mRNA vaccination in a nationwide setting. Ann Acad Med Singap 51: 96-100. https://doi.org/10.47102/annals-acadmedsg.2021425
|
| [126] |
Galván-Peña S, Leon J, Chowdhary K, et al. (2021) Profound Treg perturbations correlate with COVID-19 severity. Proc Natl Acad Sci USA 118. https://doi.org/10.1073/pnas.2111315118
|
| [127] |
Neumann J, Prezzemolo T, Vanderbeke L, et al. (2020) Increased IL-10-producing regulatory T cells are characteristic of severe cases of COVID-19. Clin Transl Immunol 9: e1204. https://doi.org/10.1002/cti2.1204
|
| [128] |
Vick SC, Frutoso M, Mair F, et al. (2021) A regulatory T cell signature distinguishes the immune landscape of COVID-19 patients from those with other respiratory infections. Sci Adv 7: eabj0274. https://doi.org/10.1126/sciadv.abj0274
|
| [129] |
Saheb Sharif-Askari F, Saheb Sharif-Askari N, Hafezi S, et al. (2023) Increased blood immune regulatory cells in severe COVID-19 with autoantibodies to type I interferons. Sci Rep 13: 17344. https://doi.org/10.1038/s41598-023-43675-w
|
| [130] | Wang F, Hou H, Luo Y, et al. (2020) The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight 5. https://doi.org/10.1172/jci.insight.137799 |
| [131] | Sadeghi A, Tahmasebi S, Mahmood A, et al. (2020) Th17 and Treg cells function in SARS-CoV2 patients compared with healthy controls. J Cell Physiol 4: 2829-2839. https://doi.org/10.1002/jcp.30047 |
| [132] |
Chen G, Wu D, Guo W, et al. (2020) Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 130: 2620-2629. https://doi.org/10.1172/JCI137244
|
| [133] | Mohebbi SR, Baghaei K, Rostami-Nejad M, et al. (2020) Significant changes of CD4, FOXP3, CD25, and IL6 expression level in Iranian COVID-19 patients. Gastroenterol Hepatol Bed Bench 13: 388-392. |
| [134] |
Caldrer S, Mazzi C, Bernardi M, et al. (2021) Regulatory T cells as predictors of clinical course in hospitalised COVID-19 patients. Front Immunol 12: 789735. https://doi.org/10.3389/fimmu.2021.789735
|
| [135] |
Meckiff BJ, Ramírez-Suástegui C, Fajardo V, et al. (2020) Imbalance of regulatory and cytotoxic SARS-CoV-2-Reactive CD4(+) T Cells in COVID-19. Cell 183: 1340-1353.e16. https://doi.org/10.3410/f.738790055.793579433
|
| [136] |
Rojas M, Herrán M, Ramírez-Santana C, et al. (2023) Molecular mimicry and autoimmunity in the time of COVID-19. J Autoimmun 139: 103070. https://doi.org/10.1016/j.jaut.2023.103070
|
| [137] |
Gouttefangeas C, Klein R, Maia A (2023) The good and the bad of T cell cross-reactivity: challenges and opportunities for novel therapeutics in autoimmunity and cancer. Front Immunol 14. https://doi.org/10.3389/fimmu.2023.1212546
|
| [138] |
Grassi F, Salina G (2023) The P2X7 receptor in autoimmunity. Int J Mol Sci 24: 14116. https://doi.org/10.3390/ijms241814116
|
| [139] |
Di Virgilio F, Giuliani AL (2016) Purinergic signalling in autoimmunity: A role for the P2X7R in systemic lupus erythematosus?. Biomed J 39: 326-338. https://doi.org/10.1016/j.bj.2016.08.006
|
| [140] |
Drury AN, Szent-Gyorgyi A (1929) The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol 68: 213-237. https://doi.org/10.1113/jphysiol.1929.sp002608
|
| [141] | Lohmann K (1929) Über die Pyrophosphatfraktion im Muskel. Naturwissenschaften 17: 624-625. https://doi.org/10.1007/BF01506215 |
| [142] |
Engelhardt WA, Ljubimowa MN (1939) Myosine and Adenosinetriphosphatase. Nature 144: 668-669. https://doi.org/10.1038/144668b0
|
| [143] |
Lipmann F (1940) A phosphorylated oxygenation product of pyruvic acid. J Biol Chem 134: 463-464. https://doi.org/10.1016/S0021-9258(18)73290-0
|
| [144] |
Lipmann F (1944) Enzymatic synthesis of acetyl phosphate. J Biol Chem 155: 55-70. https://doi.org/10.1016/S0021-9258(18)43172-9
|
| [145] |
Lipmann F, Jones ME, Black S, et al. (1953) The mechanism of the ATP-CoA-acetate reaction. J Cell Physiol Suppl 41: 109-112. https://doi.org/10.1002/jcp.1030410408
|
| [146] |
Feldberg W, Hebb C (1948) The stimulating action of phosphate compounds on the perfused superior cervical ganglion of the cat. J Physiol 107: 210-221. https://doi.org/10.1113/jphysiol.1948.sp004264
|
| [147] |
Holton P (1959) The liberation of adenosine triphosphate on antidromic stimulation of sensory nerves. J Physiol 145: 494-504. https://doi.org/10.1113/jphysiol.1959.sp006157
|
| [148] | Burnstock G (1972) Purinergic nerves. Pharmacol Rev 24: 509-581. |
| [149] |
Burnstock G (2012) Purinergic signalling: Its unpopular beginning, its acceptance and its exciting future. Bioessays 34: 218-225. https://doi.org/10.1002/bies.201100130
|
| [150] |
Burnstock G (2014) Purinergic signalling: from discovery to current developments. Exp Physiol 99: 16-34. https://doi.org/10.1113/expphysiol.2013.071951
|
| [151] |
Surprenant A, Rassendren F, Kawashima E, et al. (1996) The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272: 735-738. https://doi.org/10.1126/science.272.5262.735
|
| [152] |
Di Virgilio F (1995) The P2Z purinoceptor: an intriguing role in immunity, inflammation and cell death. Immunol Today 16: 524-528. https://doi.org/10.1016/0167-5699(95)80045-X
|
| [153] |
Ai Y, Wang H, Liu L, et al. (2023) Purine and purinergic receptors in health and disease. MedComm 4: e359. https://doi.org/10.1002/mco2.359
|
| [154] |
Burnstock G (2020) Introduction to purinergic signaling. Methods Mol Biol 2041: 1-15. https://doi.org/10.1007/978-1-4939-9717-6_1
|
| [155] | Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50: 413-492. |
| [156] |
Burnstock G (2018) Purine and purinergic receptors. Brain Neurosci Adv 2. https://doi.org/10.1177/2398212818817494
|
| [157] |
Fredholm BB, Frenguelli BG, Hills R, et al. (2023) Adenosine receptors in GtoPdb v.2023.1. IUPHAR/BPS Guide to Pharmacology CITE 2023. https://doi.org/10.2218/gtopdb/F3/2023.1
|
| [158] | Di Virgilio F, Falzoni S, Fortuny-Gomez A, et al. (2023) P2X receptors in GtoPdb v.2023.1. IUPHAR/BPS Guide to Pharmacology CITE . https://doi.org/10.2218/gtopdb/F77/2023.1 |
| [159] | Abbracchio M-P, Boeynaems J-M, Boyer JL, et al. (2023) P2Y receptors in GtoPdb v.2023.1. IUPHAR/BPS Guide to Pharmacology CITE 1. https://doi.org/10.2218/gtopdb/F52/2023.1 |
| [160] |
Cotrina ML, Nedergaard M (2009) Physiological and pathological functions of P2X7 receptor in the spinal cord. Purinergic Signal 5: 223-232. https://doi.org/10.1007/s11302-009-9138-2
|
| [161] |
Alves LA, Bezerra RJ, Faria RX, et al. (2013) Physiological roles and potential therapeutic applications of the P2X7 receptor in inflammation and pain. Molecules 18: 10953-10972. https://doi.org/10.3390/molecules180910953
|
| [162] |
Hasan D, Blankman P, Nieman GF (2017) Purinergic signalling links mechanical breath profile and alveolar mechanics with the pro-inflammatory innate immune response causing ventilation-induced lung injury. Purinergic Signal 13: 363-386. https://doi.org/10.1007/s11302-017-9564-5
|
| [163] |
Hasan D, Satalin J, van der Zee P, et al. (2018) Excessive extracellular ATP desensitizes P2Y2 and P2X4 ATP receptors provoking surfactant impairment ending in ventilation-induced lung injury. Int J Mol Sci 19: 1185. https://doi.org/10.3390/ijms19041185
|
| [164] |
Patel AS, Reigada D, Mitchell CH, et al. (2005) Paracrine stimulation of surfactant secretion by extracellular ATP in response to mechanical deformation. Am J Physiol Lung Cell MolPhysiol 289: L489-L496. https://doi.org/10.1152/ajplung.00074.2005
|
| [165] |
Kim KC, Zheng QX, Van-Seuningen I (1993) Involvement of a signal transduction mechanism in ATP-induced mucin release from cultured airway goblet cells. Am J Respir Cell Mol Biol 8: 121-125. https://doi.org/10.1165/ajrcmb/8.2.121
|
| [166] |
Tozzi M, Larsen AT, Lange SC, et al. (2018) The P2X7 receptor and pannexin-1 are involved in glucose-induced autocrine regulation in beta-cells. Sci Rep 8: 8926. https://doi.org/10.1038/s41598-018-27281-9
|
| [167] |
Jacques-Silva MC, Correa-Medina M, Cabrera O, et al. (2010) ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic beta cell. Proc Natl Acad Sci USA 107: 6465-6470. https://doi.org/10.1073/pnas.0908935107
|
| [168] |
Eltzschig HK, Sitkovsky MV, Robson SC (2012) Purinergic signaling during inflammation. N Engl J Med 367: 2322-2333. https://doi.org/10.1056/NEJMra1205750
|
| [169] |
Idzko M, Ferrari D, Eltzschig HK (2014) Nucleotide signalling during inflammation. Nature 509: 310-317. https://doi.org/10.1038/nature13085
|
| [170] |
Cekic C, Linden J (2016) Purinergic regulation of the immune system. Nat Rev Immunol 16: 177-192. https://doi.org/10.1038/nri.2016.4
|
| [171] |
Adinolfi E, Giuliani AL, De Marchi E, et al. (2018) The P2X7 receptor: A main player in inflammation. Biochem Pharmacol 151: 234-244. https://doi.org/10.1016/j.bcp.2017.12.021
|
| [172] |
Savio LEB, de Andrade Mello P, da Silva CG, et al. (2018) The P2X7 receptor in inflammatory diseases: Angel or demon?. Front Pharmacol 9: 52. https://doi.org/10.3389/fphar.2018.00052
|
| [173] |
Zarek PE, Huang CT, Lutz ER, et al. (2008) A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 111: 251-259. https://doi.org/10.1182/blood-2007-03-081646
|
| [174] |
Ohta A, Kini R, Ohta A, et al. (2012) The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol 3: 190. https://doi.org/10.3389/fimmu.2012.00190
|
| [175] | Shi L, Feng M, Du S, et al. (2019) Adenosine generated by regulatory T cells induces CD8(+) T cell exhaustion in gastric cancer through A2aR pathway. Biomed Res Int 2019: 4093214. https://doi.org/10.1155/2019/4093214 |
| [176] |
Wiley JS, Gu BJ (2012) A new role for the P2X7 receptor: a scavenger receptor for bacteria and apoptotic cells in the absence of serum and extracellular ATP. Purinergic Signal 8: 579-586. https://doi.org/10.1007/s11302-012-9308-5
|
| [177] |
Gu BJ, Saunders BM, Petrou S, et al. (2011) P2X(7) is a scavenger receptor for apoptotic cells in the absence of its ligand, extracellular ATP. J Immunol 187: 2365-2375. https://doi.org/10.4049/jimmunol.1101178
|
| [178] |
Brandao-Burch A, Key ML, Patel JJ, et al. (2012) The P2X7 receptor is an important regulator of extracellular ATP levels. Front Endocrinol (Lausanne) 3: 41. https://doi.org/10.3389/fendo.2012.00041
|
| [179] |
Gutierrez-Martin Y, Bustillo D, Gomez-Villafuertes R, et al. (2011) P2X7 receptors trigger ATP exocytosis and modify secretory vesicle dynamics in neuroblastoma cells. J Biol Chem 286: 11370-11381. https://doi.org/10.1074/jbc.M110.139410
|
| [180] |
Suadicani SO, Brosnan CF, Scemes E (2006) P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J Neurosci 26: 1378-1385. https://doi.org/10.1523/JNEUROSCI.3902-05.2006
|
| [181] |
Henriquez M, Herrera-Molina R, Valdivia A, et al. (2011) ATP release due to Thy-1-integrin binding induces P2X7-mediated calcium entry required for focal adhesion formation. J Cell Sci 124: 1581-1588. https://doi.org/10.1242/jcs.073171
|
| [182] |
Mishra A, Chintagari NR, Guo Y, et al. (2011) Purinergic P2X7 receptor regulates lung surfactant secretion in a paracrine manner. J Cell Sci 124: 657-668. https://doi.org/10.1242/jcs.066977
|
| [183] |
Ohshima Y, Tsukimoto M, Takenouchi T, et al. (2010) gamma-Irradiation induces P2X(7) receptor-dependent ATP release from B16 melanoma cells. Biochim Biophys Acta 1800: 40-46.
|
| [184] |
Johnsen B, Kaschubowski KE, Nader S, et al. (2019) P2X7A-mediated ATP secretion is accompanied by depletion of cytosolic ATP. Purinergic Signal 15: 155-166. https://doi.org/10.1007/s11302-019-09654-5
|
| [185] | Sáez PJ, Vargas P, Shoji KF, et al. (2017) ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X(7) receptors. Sci Signal 10. https://doi.org/10.1126/scisignal.aah7107 |
| [186] | Basu M, Gupta P, Dutta A, et al. (2020) Increased host ATP efflux and its conversion to extracellular adenosine is crucial for establishing Leishmania infection. J Cell Sci 133. https://doi.org/10.1242/jcs.239939 |
| [187] |
Atkinson SK, Morice AH, Sadofsky LR (2020) Rhinovirus-16 increases ATP release in A549 cells without concomitant increase in production. ERJ Open Res 6. https://doi.org/10.1183/23120541.00159-2020
|
| [188] | Ledderose C, Valsami EA, Elevado M, et al. (2023) ATP release from influenza-infected lungs enhances neutrophil activation and promotes disease progression. J Infect Dis . https://doi.org/10.1093/infdis/jiad442 |
| [189] |
Zhang C, He H, Wang L, et al. (2017) Virus-triggered ATP release limits viral replication through facilitating IFN-β production in a P2X7-dependent manner. J Immunol 199: 1372-1381. https://doi.org/10.4049/jimmunol.1700187
|
| [190] |
Adinolfi E, Callegari MG, Ferrari D, et al. (2005) Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol Biol Cell 16: 3260-3272. https://doi.org/10.1091/mbc.e04-11-1025
|
| [191] |
Ghazi K, Deng-Pichon U, Warnet JM, et al. (2012) Hyaluronan fragments improve wound healing on in vitro cutaneous model through P2X7 purinoreceptor basal activation: role of molecular weight. PLoS One 7: e48351. https://doi.org/10.1371/journal.pone.0048351
|
| [192] |
Adinolfi E, Callegari MG, Cirillo M, et al. (2009) Expression of the P2X7 receptor increases the Ca2+ content of the endoplasmic reticulum, activates NFATc1, and protects from apoptosis. J Biol Chem 284: 10120-10128. https://doi.org/10.1074/jbc.M805805200
|
| [193] |
Gilbert SM, Oliphant CJ, Hassan S, et al. (2019) ATP in the tumour microenvironment drives expression of nfP2X7, a key mediator of cancer cell survival. Oncogene 38: 194-208. https://doi.org/10.1038/s41388-018-0426-6
|
| [194] |
Pellegatti P, Falzoni S, Donvito G, et al. (2011) P2X7 receptor drives osteoclast fusion by increasing the extracellular adenosine concentration. Faseb j 25: 1264-1274. https://doi.org/10.1096/fj.10-169854
|
| [195] |
Amoroso F, Falzoni S, Adinolfi E, et al. (2012) The P2X7 receptor is a key modulator of aerobic glycolysis. Cell Death Dis 3: e370. https://doi.org/10.1038/cddis.2012.105
|
| [196] |
Schenk U, Frascoli M, Proietti M, et al. (2011) ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci Signal 4: ra12. https://doi.org/10.1126/scisignal.2001270
|
| [197] |
Figliuolo VR, Savio LEB, Safya H, et al. (2017) P2X7 receptor promotes intestinal inflammation in chemically induced colitis and triggers death of mucosal regulatory T cells. Biochim Biophys Acta Mol Basis Dis 1863: 1183-1194. https://doi.org/10.1016/j.bbadis.2017.03.004
|
| [198] |
Koo TY, Lee JG, Yan JJ, et al. (2017) The P2X7 receptor antagonist, oxidized adenosine triphosphate, ameliorates renal ischemia-reperfusion injury by expansion of regulatory T cells. Kidney Int 92: 415-431. https://doi.org/10.1016/j.kint.2017.01.031
|
| [199] |
Hubert S, Rissiek B, Klages K, et al. (2010) Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. J Exp Med 207: 2561-2568. https://doi.org/10.1084/jem.20091154
|
| [200] |
Wilhelm K, Ganesan J, Muller T, et al. (2010) Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med 16: 1434-1438. https://doi.org/10.1038/nm.2242
|
| [201] |
Elhage A, Cuthbertson P, Sligar C, et al. (2023) A species-specific anti-human P2X7 monoclonal antibody reduces Graft-versus-Host disease in humanised mice. Pharmaceutics 15. https://doi.org/10.3390/pharmaceutics15092263
|
| [202] |
Raffaghello L, Principi E, Baratto S, et al. (2022) P2X7 receptor antagonist reduces fibrosis and inflammation in a mouse model of alpha-sarcoglycan muscular dystrophy. Pharmaceuticals (Basel) 15. https://doi.org/10.3390/ph15010089
|
| [203] |
Pupovac A, Foster CM, Sluyter R (2013) Human P2X7 receptor activation induces the rapid shedding of CXCL16. Biochem Biophys Res Commun 432: 626-631. https://doi.org/10.1016/j.bbrc.2013.01.134
|
| [204] |
Pupovac A, Geraghty NJ, Watson D, et al. (2015) Activation of the P2X7 receptor induces the rapid shedding of CD23 from human and murine B cells. Immunol Cell Biol 93: 77-85. https://doi.org/10.1038/icb.2014.69
|
| [205] |
Yip L, Woehrle T, Corriden R, et al. (2009) Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J 23: 1685-1693. https://doi.org/10.1096/fj.08-126458
|
| [206] |
Huang SW, Walker C, Pennock J, et al. (2017) P2X7 receptor-dependent tuning of gut epithelial responses to infection. Immunol Cell Biol 95: 178-188. https://doi.org/10.1038/icb.2016.75
|
| [207] |
Gu BJ, Wiley JS (2006) Rapid ATP-induced release of matrix metalloproteinase 9 is mediated by the P2X7 receptor. Blood 107: 4946-4953. https://doi.org/10.1182/blood-2005-07-2994
|
| [208] |
de Torre-Minguela C, Barbera-Cremades M, Gomez AI, et al. (2016) Macrophage activation and polarization modify P2X7 receptor secretome influencing the inflammatory process. Sci Rep 6: 22586. https://doi.org/10.1038/srep22586
|
| [209] |
Csoka B, Nemeth ZH, Toro G, et al. (2015) Extracellular ATP protects against sepsis through macrophage P2X7 purinergic receptors by enhancing intracellular bacterial killing. Faseb j 29: 3626-3637. https://doi.org/10.1096/fj.15-272450
|
| [210] |
Wareham KJ, Seward EP (2016) P2X7 receptors induce degranulation in human mast cells. Purinergic Signal 12: 235-246. https://doi.org/10.1007/s11302-016-9497-4
|
| [211] |
Kawamura H, Aswad F, Minagawa M, et al. (2006) P2X7 receptors regulate NKT cells in autoimmune hepatitis. J Immunol 176: 2152-2160. https://doi.org/10.4049/jimmunol.176.4.2152
|
| [212] |
Martel-Gallegos G, Rosales-Saavedra MT, Reyes JP, et al. (2010) Human neutrophils do not express purinergic P2X7 receptors. Purinergic Signal 6: 297-306. https://doi.org/10.1007/s11302-010-9178-7
|
| [213] |
Pelegrin P, Surprenant A (2006) Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. Embo j 25: 5071-5082. https://doi.org/10.1038/sj.emboj.7601378
|
| [214] |
Suh BC, Kim JS, Namgung U, et al. (2001) P2X7 nucleotide receptor mediation of membrane pore formation and superoxide generation in human promyelocytes and neutrophils. J Immunol 166: 6754-6763. https://doi.org/10.4049/jimmunol.166.11.6754
|
| [215] |
Tsukimoto M, Maehata M, Harada H, et al. (2006) P2X7 receptor-dependent cell death is modulated during murine T cell maturation and mediated by dual signaling pathways. J Immunol 177: 2842-2850. https://doi.org/10.4049/jimmunol.177.5.2842
|
| [216] |
Aswad F, Dennert G (2006) P2X7 receptor expression levels determine lethal effects of a purine based danger signal in T lymphocytes. Cell Immunol 243: 58-65. https://doi.org/10.1016/j.cellimm.2006.12.003
|
| [217] |
Scheuplein F, Schwarz N, Adriouch S, et al. (2009) NAD+ and ATP released from injured cells induce P2X7-dependent shedding of CD62L and externalization of phosphatidylserine by murine T cells. J Immunol 182: 2898-2908. https://doi.org/10.4049/jimmunol.0801711
|
| [218] |
Albayati S, Vemulapalli H, Tsygankov AY, et al. (2021) P2Y(12) antagonism results in altered interactions between platelets and regulatory T cells during sepsis. J Leukoc Biol 110: 141-153. https://doi.org/10.1002/JLB.3A0220-097R
|
| [219] |
Vemulapalli H, Samara A, Tsygankov AY, et al. (2018) Blockade of the P2Y12 signaling pathway with clopidogrel restrains treg proliferation in vivo during sepsis and in Vitro. Blood 132: 2413. https://doi.org/10.1182/blood-2018-99-114884
|
| [220] |
Minns MS, Trinkaus-Randall V (2016) Purinergic signaling in corneal wound healing: A tale of 2 receptors. J Ocul Pharmacol Ther 32: 498-503. https://doi.org/10.1089/jop.2016.0009
|
| [221] |
Hwang SM, Koo NY, Choi SY, et al. (2009) P2X7 receptor-mediated membrane blebbing in salivary epithelial cells. Korean J Physiol Pharmacol 13: 175-179. https://doi.org/10.4196/kjpp.2009.13.3.175
|
| [222] |
Gu BJ, Wiley JS (2018) P2X7 as a scavenger receptor for innate phagocytosis in the brain. Br J Pharmacol 175: 4195-4208. https://doi.org/10.1111/bph.14470
|
| [223] |
Okura D, Horishita T, Ueno S, et al. (2015) Lidocaine preferentially inhibits the function of purinergic P2X7 receptors expressed in Xenopus oocytes. Anesth Analg 120: 597-605. https://doi.org/10.1213/ANE.0000000000000585
|
| [224] |
North RA (2016) P2X receptors. Philos Trans R Soc Lond B Biol Sci 371. https://doi.org/10.1098/rstb.2015.0427
|
| [225] |
Coddou C, Stojilkovic SS, Huidobro-Toro JP (2011) Allosteric modulation of ATP-gated P2X receptor channels. Rev Neurosci 22: 335-354. https://doi.org/10.1515/rns.2011.014
|
| [226] |
Coddou C, Yan Z, Obsil T, et al. (2011) Activation and regulation of purinergic P2X receptor channels. Pharmacol Rev 63: 641-683. https://doi.org/10.1124/pr.110.003129
|
| [227] |
Sanabria P, Ross E, Ramirez E, et al. (2008) P2Y2 receptor desensitization on single endothelial cells. Endothelium 15: 43-51. https://doi.org/10.1080/10623320802092294
|
| [228] |
Schulte G, Fredholm BB (2000) Human adenosine A(1), A(2A), A(2B), and A(3) receptors expressed in Chinese hamster ovary cells all mediate the phosphorylation of extracellular-regulated kinase 1/2. Mol Pharmacol 58: 477-482. https://doi.org/10.1124/mol.58.3.477
|
| [229] |
Klaasse EC, Ijzerman AP, de Grip WJ, et al. (2008) Internalization and desensitization of adenosine receptors. Purinergic Signal 4: 21-37. https://doi.org/10.1007/s11302-007-9086-7
|
| [230] |
Illes P, Müller CE, Jacobson KA, et al. (2021) Update of P2X receptor properties and their pharmacology: IUPHAR Review 30. Br J Pharmacol 178: 489-514. https://doi.org/10.1111/bph.15299
|
| [231] |
Jacobson KA, Delicado EG, Gachet C, et al. (2020) Update of P2Y receptor pharmacology: IUPHAR Review 27. Br J Pharmacol 177: 2413-2433. https://doi.org/10.1111/bph.15005
|
| [232] |
Jacobson KA, Muller CE (2016) Medicinal chemistry of adenosine, P2Y and P2X receptors. Neuropharmacology 104: 31-49. https://doi.org/10.1016/j.neuropharm.2015.12.001
|
| [233] |
Myers AJ, Eilertson B, Fulton SA, et al. (2005) The purinergic P2X7 receptor Is not required for control of pulmonary Mycobacterium tuberculosis infection. Infect Immun 73: 3192-3195. https://doi.org/10.1128/IAI.73.5.3192-3195.2005
|
| [234] |
Santiago-Carvalho I, de Almeida-Santos G, Bomfim CCB, et al. (2021) P2x7 receptor signaling blockade reduces lung inflammation and necrosis during severe experimental tuberculosis. Front Cell Infect Microbiol 11: 672472. https://doi.org/10.3389/fcimb.2021.672472
|
| [235] |
Ardissone V, Radaelli E, Zaratin P, et al. (2011) Pharmacologic P2X purinergic receptor antagonism in the treatment of collagen-induced arthritis. Arthritis Rheum 63: 3323-3332. https://doi.org/10.1002/art.30556
|
| [236] |
Martinez CG, Zamith-Miranda D, da Silva MG, et al. (2015) P2×7 purinergic signaling in dilated cardiomyopathy induced by auto-immunity against muscarinic M2 receptors: autoantibody levels, heart functionality and cytokine expression. Sci Rep 5: 16940. https://doi.org/10.1038/srep16940
|
| [237] |
Baldini C, Santini E, Rossi C, et al. (2017) The P2X7 receptor-NLRP3 inflammasome complex predicts the development of non-Hodgkin's lymphoma in Sjogren's syndrome: a prospective, observational, single-centre study. J Intern Med 282: 175-186. https://doi.org/10.1111/joim.12631
|
| [238] |
Baldini C, Rossi C, Ferro F, et al. (2013) The P2X7 receptor-inflammasome complex has a role in modulating the inflammatory response in primary Sjögren's syndrome. J Intern Med 274: 480-489. https://doi.org/10.1111/joim.12115
|
| [239] |
Di Virgilio F, Bronte V, Collavo D, et al. (1989) Responses of mouse lymphocytes to extracellular adenosine 5′-triphosphate (ATP). Lymphocytes with cytotoxic activity are resistant to the permeabilizing effects of ATP. J Immunol 143: 1955-1960. https://doi.org/10.4049/jimmunol.143.6.1955
|
| [240] |
Filippini A, Taffs RE, Agui T, et al. (1990) Ecto-ATPase activity in cytolytic T-lymphocytes. Protection from the cytolytic effects of extracellular ATP. J Biol Chem 265: 334-340. https://doi.org/10.1016/S0021-9258(19)40234-2
|
| [241] |
Falcone C, Caracciolo M, Correale P, et al. (2020) Can adenosine fight COVID-19 acute respiratory distress syndrome?. J Clin Med 9. https://doi.org/10.20944/preprints202007.0426.v1
|
| [242] |
Fajgenbaum DC, June CH (2020) Cytokine Storm. N Engl J Med 383: 2255-2273. https://doi.org/10.1056/NEJMra2026131
|
| [243] |
Young CNJ, Chira N, Róg J, et al. (2018) Sustained activation of P2X7 induces MMP-2-evoked cleavage and functional purinoceptor inhibition. J Mol Cell Biol 10: 229-242. https://doi.org/10.1093/jmcb/mjx030
|
| [244] |
Young CNJ, Górecki DC (2018) P2RX7 purinoceptor as a therapeutic target-the second coming?. Front Chem 6: 248. https://doi.org/10.3389/fchem.2018.00248
|
| [245] |
Zhao R, Qiao J, Zhang X, et al. (2019) Toll-like receptor-mediated activation of CD39 internalization in BMDCs leads to extracellular ATP accumulation and facilitates P2X7 receptor activation. Front Immunol 10: 2524. https://doi.org/10.3389/fimmu.2019.02524
|
| [246] |
Hotchkiss RS, Monneret G, Payen D (2013) Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 13: 260-268. https://doi.org/10.1016/S1473-3099(13)70001-X
|
| [247] |
Hotchkiss RS, Monneret G, Payen D (2013) Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13: 862-874. https://doi.org/10.1038/nri3552
|
| [248] |
Ward NS, Casserly B, Ayala A (2008) The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med 29: 617-625. https://doi.org/10.1016/j.ccm.2008.06.010
|
| [249] |
Bone RC (1996) Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med 24: 1125-1128. https://doi.org/10.1097/00003246-199607000-00010
|
| [250] |
Biscetti L, De Vanna G, Cresta E, et al. (2021) Headache and immunological/autoimmune disorders: a comprehensive review of available epidemiological evidence with insights on potential underlying mechanisms. J Neuroinflammation 18: 259. https://doi.org/10.1186/s12974-021-02229-5
|
| [251] |
John S, Hajj-Ali RA (2014) Headache in autoimmune diseases. Headache 54: 572-582. https://doi.org/10.1111/head.12306
|
| [252] |
Kim HJ (2021) Pruritus in autoimmune connective tissue diseases. Ann Transl Med 9: 441. https://doi.org/10.21037/atm-20-4894
|
| [253] |
Zielinski MR, Systrom DM, Rose NR (2019) Fatigue, sleep, and autoimmune and related disorders. Front Immunol 10: 1827. https://doi.org/10.3389/fimmu.2019.01827
|
| [254] |
Morris G, Berk M, Walder K, et al. (2015) Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses. BMC Med 13: 28. https://doi.org/10.1186/s12916-014-0259-2
|
| [255] |
Mifflin KA, Kerr BJ (2017) Pain in autoimmune disorders. J Neurosci Res 95: 1282-1294. https://doi.org/10.1002/jnr.23844
|
| [256] |
Lacagnina MJ, Heijnen CJ, Watkins LR, et al. (2021) Autoimmune regulation of chronic pain. Pain Rep 6: e905. https://doi.org/10.1097/PR9.0000000000000905
|
| [257] |
Joaquim AF, Appenzeller S (2015) Neuropsychiatric manifestations in rheumatoid arthritis. Autoimmun Rev 14: 1116-1122. https://doi.org/10.1016/j.autrev.2015.07.015
|
| [258] |
Shim CH, Cho S, Shin YM, et al. (2022) Emerging role of bystander T cell activation in autoimmune diseases. BMB Rep 55: 57-64. https://doi.org/10.5483/BMBRep.2022.55.2.183
|
| [259] |
Fujinami RS, von Herrath MG, Christen U, et al. (2006) Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev 19: 80-94. https://doi.org/10.1128/CMR.19.1.80-94.2006
|
| [260] |
van Aalst S, Ludwig IS, van der Zee R, et al. (2017) Bystander activation of irrelevant CD4+ T cells following antigen-specific vaccination occurs in the presence and absence of adjuvant. PLoS One 12: e0177365. https://doi.org/10.1371/journal.pone.0177365
|
| [261] |
Bergamaschi L, Mescia F, Turner L, et al. (2021) Longitudinal analysis reveals that delayed bystander CD8+ T cell activation and early immune pathology distinguish severe COVID-19 from mild disease. Immunity 54: 1257-1275.e1258. https://doi.org/10.1016/j.immuni.2021.05.010
|
| [262] |
Lindenbergh MFS, Koerhuis DGJ, Borg EGF, et al. (2019) Bystander T-Cells support clonal T-cell activation by controlling the release of dendritic cell-derived immune-stimulatory extracellular vesicles. Front Immunol 10: 448. https://doi.org/10.3389/fimmu.2019.00448
|
| [263] |
Wang JY, Zhang W, Roehrl VB, et al. (2022) An autoantigen atlas from human lung HFL1 cells offers clues to neurological and diverse autoimmune manifestations of COVID-19. Front Immunol 13: 831849. https://doi.org/10.3389/fimmu.2022.831849
|
| [264] |
Felipe Cuspoca A, Isaac Estrada P, Velez-van-Meerbeke A (2022) Molecular mimicry of SARS-CoV-2 spike protein in the nervous system: A bioinformatics approach. Comput Struct Biotechnol J 20: 6041-6054. https://doi.org/10.1016/j.csbj.2022.10.022
|
| [265] |
Woodruff MC, Ramonell RP, Haddad NS, et al. (2022) Dysregulated naive B cells and de novo autoreactivity in severe COVID-19. Nature 611: 139-147. https://doi.org/10.1038/s41586-022-05273-0
|
| [266] |
Milne GR, Palmer TM (2011) Anti-inflammatory and immunosuppressive effects of the A2A adenosine receptor. Sci World J 11: 320-339. https://doi.org/10.1100/tsw.2011.22
|
| [267] |
Elsaghir A, El-Sabaa EMW, Ahmed AK, et al. (2023) The role of cluster of differentiation 39 (CD39) and purinergic signaling pathway in viral infections. Pathogens 12. https://doi.org/10.3390/pathogens12020279
|
| [268] |
Auclair H, Ouk-Martin C, Roland L, et al. (2019) EBV latency III-transformed B cells are inducers of conventional and unconventional regulatory T cells in a PD-L1-dependent manner. J Immunol 203: 1665-1674. https://doi.org/10.4049/jimmunol.1801420
|
| [269] | Song J-W, Huang H-H, Zhang C, et al. (2019) Expression of CD39 is correlated with HIV DNA levels in naïve tregs in chronically infected ART naïve patients. Front Immunol 10. https://doi.org/10.3389/fimmu.2019.02465 |
| [270] | Gupta PK, Godec J, Wolski D, et al. (2015) CD39 expression identifies terminally exhausted CD8+ T cells. PLoSPathog 11: e1005177. https://doi.org/10.1371/journal.ppat.1005177 |
| [271] |
Wingate PJ, McAulay KA, Anthony IC, et al. (2009) Regulatory T cell activity in primary and persistent Epstein-Barr virus infection. J Med Virol 81: 870-877. https://doi.org/10.1002/jmv.21445
|
| [272] |
Chevalier MF, Weiss L (2013) The split personality of regulatory T cells in HIV infection. Blood 121: 29-37. https://doi.org/10.1182/blood-2012-07-409755
|
| [273] |
Wang L, Qiu J, Yu L, et al. (2014) Increased numbers of CD5+CD19+CD1dhighIL-10+ Bregs, CD4+Foxp3+ Tregs, CD4+CXCR5+Foxp3+ follicular regulatory T (TFR) cells in CHB or CHC patients. J Transl Med 12: 251. https://doi.org/10.1186/s12967-014-0251-9
|
| [274] |
Cash E, Goodwin AT, Tatler AL (2023) Adenosine receptor signalling as a driver of pulmonary fibrosis. Pharmacol Ther 249: 108504. https://doi.org/10.1016/j.pharmthera.2023.108504
|
| [275] |
Fausther M (2018) Extracellular adenosine: a critical signal in liver fibrosis. Am J Physiol Gastrointest Liver Physiol 315: G12-G19. https://doi.org/10.1152/ajpgi.00006.2018
|
| [276] |
Sheng G, Chen P, Wei Y, et al. (2020) Viral infection increases the risk of idiopathic pulmonary fibrosis: A meta-analysis. Chest 157: 1175-1187. https://doi.org/10.1016/j.chest.2019.10.032
|
| [277] |
Androutsakos T, Schina M, Pouliakis A, et al. (2020) Causative factors of liver fibrosis in HIV-infected patients. A single center study. BMC Gastroenterol 20: 91. https://doi.org/10.1186/s12876-020-01230-1
|
| [278] |
McCaughan GW, George J (2004) Fibrosis progression in chronic hepatitis C virus infection. Gut 53: 318-321. https://doi.org/10.1136/gut.2003.026393
|
| [279] |
Rockey DC, Friedman SL (2021) Fibrosis regression after eradication of hepatitis C virus: From bench to bedside. Gastroenterology 160: 1502-1520.e1501. https://doi.org/10.1053/j.gastro.2020.09.065
|
| [280] |
Stock TC, Bloom BJ, Wei N, et al. (2012) Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J Rheumatol 39: 720-727. https://doi.org/10.3899/jrheum.110874
|
| [281] |
Keystone EC, Wang MM, Layton M, et al. (2012) Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann Rheum Dis 71: 1630-1635. https://doi.org/10.1136/annrheumdis-2011-143578
|
| [282] |
Recourt K, de Boer P, van der Ark P, et al. (2023) Characterization of the central nervous system penetrant and selective purine P2X7 receptor antagonist JNJ-54175446 in patients with major depressive disorder. Transl Psychiatry 13: 266. https://doi.org/10.1038/s41398-023-02557-5
|
| [283] |
Tamargo J, Le Heuzey JY, Mabo P (2015) Narrow therapeutic index drugs: a clinical pharmacological consideration to flecainide. Eur J Clin Pharmacol 71: 549-567. https://doi.org/10.1007/s00228-015-1832-0
|
| [284] | Eser A, Colombel JF, Rutgeerts P, et al. (2015) Safety and efficacy of an oral inhibitor of the purinergic receptor P2X7 in adult patients with moderately to severely active crohn's disease: A randomized placebo-controlled, double-blind, phase IIa study. Inflamm Bowel Dis 21: 2247-2253. https://doi.org/10.1097/MIB.0000000000000514 |
| [285] |
Neves AR, Castelo-Branco MT, Figliuolo VR, et al. (2014) Overexpression of ATP-activated P2X7 receptors in the intestinal mucosa is implicated in the pathogenesis of Crohn's disease. Inflamm Bowel Dis 20: 444-457. https://doi.org/10.1097/01.MIB.0000441201.10454.06
|
Figures(4) / Tables(2)

Djo Hasan. Purinergic P2X7R expressed on regulatory T cells potentially links molecular mimicry to autoimmune responses[J]. AIMS Allergy and Immunology, 2024, 8(2): 80-123. doi: 10.3934/Allergy.2024006







 DownLoad:
DownLoad: