Citation: Kenta Yamauchi, Shinji Kondo, Makiko Hamamoto, Yutaka Suzuki, Hiromi Nishida. Genome-wide maps of nucleosomes of the trichostatin A treated and untreated archiascomycetous yeast Saitoella complicata[J]. AIMS Microbiology, 2016, 2(1): 69-91. doi: 10.3934/microbiol.2016.1.69

| [1] |
Igo-Kemenes T, Hörz W, Zachau HG (1982) Chromatin. Ann Rev Biochem 51: 89–121. doi: 10.1146/annurev.bi.51.070182.000513

|
| [2] | Luger K, Mäder AW, Richmond RK, et al. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389: 251–260. |
| [3] |
Millar CB, Grunstein M (2006) Genome-wide patterns of histone modifications in yeast. Nat Rev Mol Cell Biol 7: 657–666. doi: 10.1038/nrm1986
|
| [4] |
Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128: 707–719. doi: 10.1016/j.cell.2007.01.015
|
| [5] |
Luger K, Richmond TJ (1998) The histone tails of the nucleosome. Curr Opin Genet Dev 8: 140–146. doi: 10.1016/S0959-437X(98)80134-2
|
| [6] |
Sasaki K, Ito T, Nishino N, et al. (2009) Real-time imaging of histone H4 hyperacetylation in living cells. Proc Natl Acad Sci USA 106: 16257–16262. doi: 10.1073/pnas.0902150106
|
| [7] |
Yoshida M, Horinouchi S, Beppu T (1995) Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. BioEssays 17: 423–430. doi: 10.1002/bies.950170510
|
| [8] |
Nishida H, Motoyama T, Suzuki Y, et al. (2010) Genome-wide maps of mononucleosomes and dinucleosomes containing hyperacetylated histones of Aspergillus fumigatus. PLOS ONE 5: e9916. doi: 10.1371/journal.pone.0009916
|
| [9] | Liu Y, Leigh JW, Brinkmann H, et al. (2009) Phylogenomic analyses support the monophyly of Taphrinomycotina, including Schizosaccharomyces fission yeasts. Mol Biol Evol 26: 27–34. |
| [10] |
Nishida H, Sugiyama J (1994) Archiascomycetes: detection of a major new lineage within the Ascomycota. Mycoscience 35: 361–366. doi: 10.1007/BF02268506
|
| [11] |
Goto S, Sugiyama J, Hamamoto M, et al. (1987) Saitoella, a new anamorph genus in the Cryptococcaceae to accommodate two Himalayan yeast isolates formerly identified as Rhodotorula glutinis. J Gen Appl Microbiol 33: 75–85. doi: 10.2323/jgam.33.75
|
| [12] |
Nishida H, Hamamoto M, Sugiyama J (2011) Draft genome sequencing of the enigmatic yeast Saitoella complicata. J Gen Appl Microbiol 57: 243–246. doi: 10.2323/jgam.57.243
|
| [13] |
Nishida H, Matsumoto T, Kondo S, et al. (2014) The early diverging ascomycetous budding yeast Saitoella complicata has three histone deacetylases belonging to the Clr6, Hos2, and Rpd3 lineages. J Gen Appl Microbiol 60: 7–12. doi: 10.2323/jgam.60.7
|
| [14] |
Ekwall K, Olsson T, Tumer BM, et al. (1997) Transient inhibitor of histone deacetylation alters the structural and functional imprint at fission yeast centromeres. Cell 91: 1021–1032. doi: 10.1016/S0092-8674(00)80492-4
|
| [15] | Yamauchi K, Kondo S, Hamamoto M, et al. (2015) Draft genome sequence of the archiascomycetous yeast Saitoella complicata. Genome Announc 3: e00220–15. |
| [16] |
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. doi: 10.1093/bioinformatics/btp120
|
| [17] |
Li H, Handsaker B, Wysoker A, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. doi: 10.1093/bioinformatics/btp352
|
| [18] |
Ogasawara O, Mashima J, Kodama Y, et al. (2013) DDBJ new system and service refactoring. Nucleic Acids Res 41: D25–D29. doi: 10.1093/nar/gks1152
|
| [19] |
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for different expression analysis of digital gene expression data. Bioinformatics 26: 139–140. doi: 10.1093/bioinformatics/btp616
|
| [20] |
Valouev A, Johnson SM, Boyd SD, et al. (2011) Determinants of nucleosome organization in primary human cells. Nature 474: 516–520. doi: 10.1038/nature10002
|
| [21] | Kaplan N, Moore IK, Fondufe-Mittendorf Y, et al. (2008) The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458: 362–366. |
| [22] |
Altschul SF, Gish W, Miller W, et al. (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. doi: 10.1016/S0022-2836(05)80360-2
|
| [23] |
Amit M, Donyo M, Hollander D, et al. (2012) Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep 1: 543–556. doi: 10.1016/j.celrep.2012.03.013
|
| [24] |
Nishida H, Katayama T, Suzuki Y, et al. (2013) Base composition and nucleosome density in exonic and intronic regions in genes of the filamentous ascomycetes Aspergillus nidulans and Aspergillus oryzae. Gene 525: 5–10. doi: 10.1016/j.gene.2013.04.077
|
| [25] | Yu J, Hu S, Wang J, et al. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296: 79–92. |
| [26] |
Nishida H, Kondo S, Matsumoto T, et al. (2012) Characteristics of nucleosomes and linker DNA regions on the genome of the basidiomycete Mixia osmundae revealed by mono- and dinucleosome mapping. Open Biol 2: 120043. doi: 10.1098/rsob.120043
|
| [27] |
Graessle S, Dangl M, Haas H, et al. (2000) Characterization of two putative histone deacetylase genes from Aspergillus nidulans. Biochim Biophys Acta 1492: 120–126. doi: 10.1016/S0167-4781(00)00093-2
|
| [28] |
Andersson R, Enroth S, Rada-Iglesias A, et al. (2009) Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res 19: 1732–1741. doi: 10.1101/gr.092353.109
|
| [29] |
Chen W, Luo L, Zhang L (2010) The organization of nucleosomes around splice sites. Nucleic Acids Res 38: 2788–2798. doi: 10.1093/nar/gkq007
|
| [30] |
Schwartz S, Ast G (2010) Chromatin density and splicing density: on the cross-talk between chromatin structure and splicing. EMBO J 29: 1629–1636. doi: 10.1038/emboj.2010.71
|
| [31] |
Schwartz S, Meshorer E, Ast G (2009) Chromatin organization marks exon–intron structure. Nat Struct Mol Biol 16: 990–995. doi: 10.1038/nsmb.1659
|
| [32] |
Tilgner H, Nikolaou C, Althammer S, et al. (2009) Nucleosome positioning as a determinant of exon recognition. Nat Struct Mol Biol 16: 996–1001. doi: 10.1038/nsmb.1658
|
| [33] |
Tsankov A, Yanagisawa Y, Rhind N, et al. (2011) Evolutionary divergence of intrinsic and trans-regulated nucleosome positioning sequences reveals plastic rules for chromatin organization. Genome Res 21: 1851–1862. doi: 10.1101/gr.122267.111
|
| [34] |
Shim YS, Choi Y, Kang K, et al. (2012) Hrp3 controls nucleosome positioning to suppress non-coding transcription in eu- and heterochromatin. EMBO J 31: 4375–4387. doi: 10.1038/emboj.2012.267
|
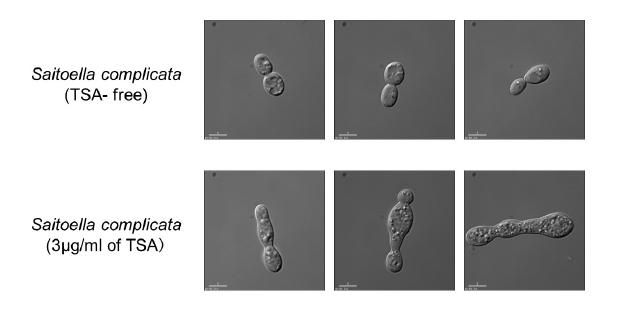
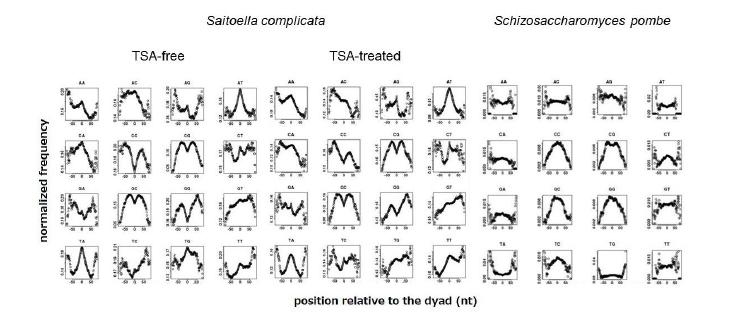
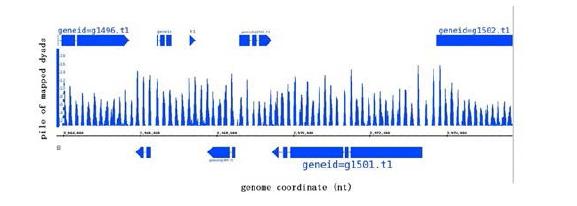
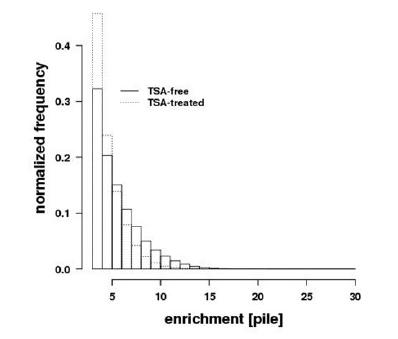
Figures(7) / Tables(2)

Kenta Yamauchi, Shinji Kondo, Makiko Hamamoto, Yutaka Suzuki, Hiromi Nishida. Genome-wide maps of nucleosomes of the trichostatin A treated and untreated archiascomycetous yeast Saitoella complicata[J]. AIMS Microbiology, 2016, 2(1): 69-91. doi: 10.3934/microbiol.2016.1.69







 DownLoad:
DownLoad: