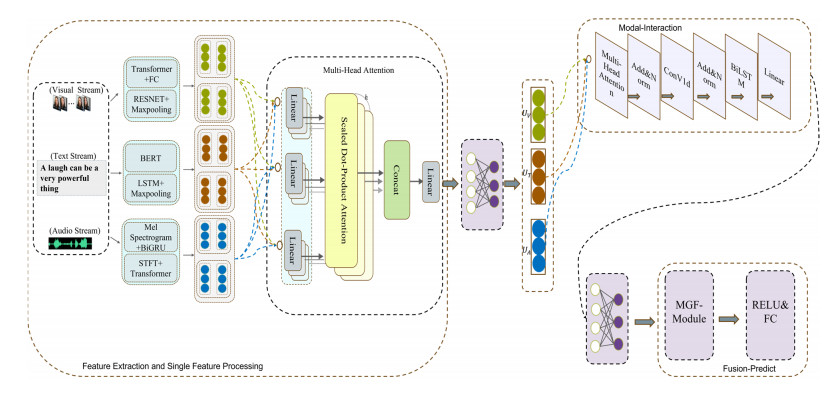
Multimodal emotion analysis involves the integration of information from various modalities to better understand human emotions. In this paper, we propose the Cross-modal Emotion Recognition based on multi-layer semantic fusion (CM-MSF) model, which aims to leverage the complementarity of important information between modalities and extract advanced features in an adaptive manner. To achieve comprehensive and rich feature extraction from multimodal sources, considering different dimensions and depth levels, we design a parallel deep learning algorithm module that focuses on extracting features from individual modalities, ensuring cost-effective alignment of extracted features. Furthermore, a cascaded cross-modal encoder module based on Bidirectional Long Short-Term Memory (BILSTM) layer and Convolutional 1D (ConV1d) is introduced to facilitate inter-modal information complementation. This module enables the seamless integration of information across modalities, effectively addressing the challenges associated with signal heterogeneity. To facilitate flexible and adaptive information selection and delivery, we design the Mask-gated Fusion Networks (MGF-module), which combines masking technology with gating structures. This approach allows for precise control over the information flow of each modality through gating vectors, mitigating issues related to low recognition accuracy and emotional misjudgment caused by complex features and noisy redundant information. The CM-MSF model underwent evaluation using the widely recognized multimodal emotion recognition datasets CMU-MOSI and CMU-MOSEI. The experimental findings illustrate the exceptional performance of the model, with binary classification accuracies of 89.1% and 88.6%, as well as F1 scores of 87.9% and 88.1% on the CMU-MOSI and CMU-MOSEI datasets, respectively. These results unequivocally validate the effectiveness of our approach in accurately recognizing and classifying emotions.
Citation: Zhijing Xu, Yang Gao. Research on cross-modal emotion recognition based on multi-layer semantic fusion[J]. Mathematical Biosciences and Engineering, 2024, 21(2): 2488-2514. doi: 10.3934/mbe.2024110
Multimodal emotion analysis involves the integration of information from various modalities to better understand human emotions. In this paper, we propose the Cross-modal Emotion Recognition based on multi-layer semantic fusion (CM-MSF) model, which aims to leverage the complementarity of important information between modalities and extract advanced features in an adaptive manner. To achieve comprehensive and rich feature extraction from multimodal sources, considering different dimensions and depth levels, we design a parallel deep learning algorithm module that focuses on extracting features from individual modalities, ensuring cost-effective alignment of extracted features. Furthermore, a cascaded cross-modal encoder module based on Bidirectional Long Short-Term Memory (BILSTM) layer and Convolutional 1D (ConV1d) is introduced to facilitate inter-modal information complementation. This module enables the seamless integration of information across modalities, effectively addressing the challenges associated with signal heterogeneity. To facilitate flexible and adaptive information selection and delivery, we design the Mask-gated Fusion Networks (MGF-module), which combines masking technology with gating structures. This approach allows for precise control over the information flow of each modality through gating vectors, mitigating issues related to low recognition accuracy and emotional misjudgment caused by complex features and noisy redundant information. The CM-MSF model underwent evaluation using the widely recognized multimodal emotion recognition datasets CMU-MOSI and CMU-MOSEI. The experimental findings illustrate the exceptional performance of the model, with binary classification accuracies of 89.1% and 88.6%, as well as F1 scores of 87.9% and 88.1% on the CMU-MOSI and CMU-MOSEI datasets, respectively. These results unequivocally validate the effectiveness of our approach in accurately recognizing and classifying emotions.

| [1] | R. K. Patra, B. Patil, T. S. Kumar, G. Shivakanth, B. M. Manjula, Machine learning based sentiment analysis and swarm intelligence, in 2023 IEEE International Conference on Integrated Circuits and Communication Systems (ICICACS), IEEE, (2023), 1–8. https://doi.org/10.1109/ICICACS57338.2023.10100262 |
| [2] |
R. Das, T. D. Singh, Multimodal sentiment analysis: A survey of methods, trends, and challenges, ACM Comput. Surv., 55 (2023), 1–38. https://doi.org/10.1145/3586075 doi: 10.1145/3586075

|
| [3] |
S. Peng, K. Chen, T. Tian, J. Chen, An autoencoder-based feature level fusion for speech emotion recognition, Digital Commun. Networks, 2022. https://doi.org/10.1016/j.dcan.2022.10.018 doi: 10.1016/j.dcan.2022.10.018
|
| [4] | S. Yoon, S. Byun, K. Jung, Multimodal speech emotion recognition using audio and text, in 2018 IEEE Spoken Language Technology Workshop (SLT), IEEE, (2018), 112–118. https://doi.org/10.1109/SLT.2018.8639583 |
| [5] |
E. Jeong, G. Kim, S. Kang, Multimodal prompt learning in emotion recognition using context and audio information, Mathematics, 11 (2023), 2908. https://doi.org/10.3390/math11132908 doi: 10.3390/math11132908
|
| [6] |
E. Batbaatar, M. Li, K. H. Ryu, Semantic-emotion neural network for emotion recognition from text, IEEE Access, 7 (2019), 111866–111878. https://doi.org/10.1109/ACCESS.2019.2934529 doi: 10.1109/ACCESS.2019.2934529
|
| [7] | A. Zadeh, M. Chen, S. Poria, E. Cambria, L. P. Morency, Tensor fusion network for multimodal sentiment analysis, in Proceedings of the 2017 Conference on Empirical Methods in Natural Language Processing, Association for Computational Linguistics, (2017), 1103–1114. https://doi.org/10.18653/v1/D17-1115 |
| [8] | Z. Liu, Y. Shen, V. B. Lakshminarasimhan, P. P. Liang, A. B. Zadeh, L. P. Morency, Efficient low-rank multimodal fusion with modality-specific factors, in Proceedings of the 56th Annual Meeting of the Association for Computational Linguistics, Association for Computational Linguistics, (2018), 2247–2256. https://doi.org/10.18653/v1/P18-1209 |
| [9] | S. Mai, H. Hu, S. Xing, Modality to modality translation: An adversarial representation learning and graph fusion network for multimodal fusion, in Proceedings of the AAAI Conference on Artificial Intelligence, AAAI Press, (2020), 164–172. https://doi.org/10.1609/aaai.v34i01.5347 |
| [10] |
B. Kratzwald, S. Ilić, M. Kraus, S. Feuerriegel, H. Prendinger, Deep learning for affective computing: Text-based emotion recognition in decision support, Decis. Support Syst., 115 (2018), 24–35. https://doi.org/10.1016/j.dss.2018.09.002 doi: 10.1016/j.dss.2018.09.002
|
| [11] | L. Zheng, L. Sun, M. Xu, H. Sun, K. Xu, Z. Wen, et al., Explainable multimodal emotion reasoning, preprint, arXiv: 2306.15401. |
| [12] | L. Sun, B. Liu, J. Tao, Z. Lian, Multimodal cross- and self-attention network for speech emotion recognition, in ICASSP 2021-2021 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), IEEE, (2021), 4275–4279. https://doi.org/10.1109/ICASSP39728.2021.9414654 |
| [13] |
X. Liu, Z. Xu, K. Huang, Multimodal emotion recognition based on cascaded multichannel and hierarchical fusion, Comput. Intell. Neurosci., 5 (2023), 9645611. https://doi.org/10.1155/2023/9645611 doi: 10.1155/2023/9645611
|
| [14] |
S. Lee, D. K. Han, H. Ko, Multimodal emotion recognition fusion analysis adapting BERT with heterogeneous feature unification, IEEE Access, 9 (2021), 94557–94572. https://doi.org/10.1109/ACCESS.2021.3092735 doi: 10.1109/ACCESS.2021.3092735
|
| [15] | P. Kumar, X. Li, Interpretable multimodal emotion recognition using facial features and physiological signals, preprint, arXiv: 2306.02845. |
| [16] | F. Lv, X. Chen, Y. Huang, L. Duan, G. Lin, Progressive modality reinforcement for human multimodal emotion recognition from unaligned multimodal sequences, in 2021 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), IEEE, (2021), 2554–2562. https://doi.org/10.1109/CVPR46437.2021.00258 |
| [17] | D. Hazarika, R. Zimmermann, S. Poria, MISA: Modality-invariant and -specific representations for multimodal sentiment analysis, in Proceedings of the 28th ACM International Conference on Multimedia, ACM, (2020), 1122–1131. https://doi.org/10.1145/3394171.3413678 |
| [18] | D. Yang, S. Huang, H. Kuang, Y. Du, L. Zhang, Disentangled representation learning for multimodal emotion recognition, in Proceedings of the 30th ACM International Conference on Multimedia (MM'22), ACM, (2022), 1642–1651. https://doi.org/10.1145/3503161.3547754 |
| [19] | H. Han, J. Yang, W. Slamu, Cascading modular multimodal cross-attention network for rumor detection, in 2023 IEEE International Conference on Control, Electronics and Computer Technology (ICCECT), IEEE, (2023), 974–980. https://doi.org/10.1109/ICCECT57938.2023.10140211 |
| [20] | S. A. M. Zaidi, S. Latif, J. Qadir, Cross-language speech emotion recognition using multimodal dual attention transformers, preprint, arXiv: 2306.13804. |
| [21] | Z. Sun, P. Sarma, W. Sethares, Y. Liang, Learning relationships between text, audio, and video via deep canonical correlation for multimodal language analysis, in Proceedings of the AAAI Conference on Artificial Intelligence, AAAI Press, (2020), 8992–8999. https://doi.org/10.1609/aaai.v34i05.6431 |
| [22] | K. Yang, H. Xu, K. Gao, CM-BERT: Cross-Modal BERT for text-audio sentiment analysis, in Proceedings of the 28th ACM International Conference on Multimedia (MM'20), ACM, (2020), 521–528. https://doi.org/10.1145/3394171.3413690 |
| [23] | H. Yang, X. Gao, J. Wu, T. Gan, N. Ding, F. Jiang, et al., Self-adaptive context and modal-interaction modeling for multimodal emotion recognition, in Findings of the Association for Computational Linguistics: ACL 2023, Association for Computational Linguistics, (2023), 6267–6281. https://doi.org/10.18653/v1/2023.findings-acl.390 |
| [24] | G. Paraskevopoulos, E. Georgiou, A. Potamianos, Mmlatch: Bottom-up top-down fusion for multimodal sentiment analysis, in ICASSP 2022-2022 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), IEEE, (2022), 4573–4577. https://doi.org/10.1109/ICASSP43922.2022.9746418 |
| [25] |
L. Zhu, Z. Zhu, C. Zhang, Y. Xu, X. Kong, Multimodal sentiment analysis based on fusion methods: A survey, Inf. Fusion, 95 (2023), 306–325. https://doi.org/10.1016/j.inffus.2023.02.028 doi: 10.1016/j.inffus.2023.02.028
|
| [26] |
S. Zhang, S. Zhang, T. Huang, W. Gao, Q. Tian, Learning affective features with a hybrid deep model for audio–visual emotion recognition, IEEE Trans. Circuits Syst. Video Technol., 28 (2018), 3030–3043. https://doi.org/10.1109/TCSVT.2017.2719043 doi: 10.1109/TCSVT.2017.2719043
|
| [27] | D. Hazarika, S. Gorantla, S. Poria, R. Zimmermann, Self-Attentive feature-level fusion for multimodal emotion detection, in 2018 IEEE Conference on Multimedia Information Processing and Retrieval (MIPR), IEEE, (2018), 196–201. https://doi.org/10.1109/MIPR.2018.00043 |
| [28] |
M. S. Hossain, G. Muhammad, Emotikon recognition using deep learning approach from audio–visual emotional big data, Inf. Fusion, 49 (2019), 69–78. https://doi.org/10.1016/j.inffus.2018.09.008 doi: 10.1016/j.inffus.2018.09.008
|
| [29] |
H. Cheng, Z. Yang, X. Zhang, Y. Yang, Multimodal sentiment analysis based on attentional temporal convolutional network and multi-layer feature fusion, IEEE Trans. Affective Comput., 14 (2023), 3149–3163. https://doi.org/10.1109/TAFFC.2023.3265653 doi: 10.1109/TAFFC.2023.3265653
|
| [30] |
S. Wang, J. Qu, Y. Zhang, Y. Zhang, Multimodal emotion recognition from EEG signals and facial expressions, IEEE Access, 11 (2023), 33061–33068. https://doi.org/10.1109/ACCESS.2023.3263670 doi: 10.1109/ACCESS.2023.3263670
|
| [31] | C. Xu, K. Shen, H. Sun, Supplementary features of BiLSTM for enhanced sequence labeling, preprint, arXiv: 2305.19928. |
| [32] |
L. Zhu, M. Xu, Y. Bao, Y. Xu, X. Kong, Deep learning for aspect-based sentiment analysis: A review, PeerJ Comput. Sci., 8 (2022), e1044. https://doi.org/10.7717/peerj-cs.1044 doi: 10.7717/peerj-cs.1044
|
| [33] | Y. H. H. Tsai, S. Bai, P. P. Liang, J. Z. Kolter, L. P. Morency, S. Ruslan, Multimodal transformer for unaligned multimodal language sequences, in Proceedings of the 57th Annual Meeting of the Association for Computational Linguistics, NIH Public Access, (2019), 6558–6569. https://doi.org/10.18653/v1/p19-1656 |
| [34] | Y. H. Tsai, S. Bai, P. P. Liang, J. Z. Kolter, L. P. Morency, R. Salakhutdinov, Multimodal transformer for unaligned multimodal language sequences, in Proceedings of the 57th Annual Meeting of the Association for Computational Linguistics, Association for Computational Linguistics, (2019), 6558–6569. https://doi.org/10.18653/v1/p19-1656 |
| [35] | D. Hazarika, R. Zimmermann, S. Poria, MISA: Modality-invariant and -specific representations for multimodal sentiment analysis, in Proceedings of the 28th ACM International Conference on Multimedia (MM'20), ACM, (2020), 1122–1131. https://doi.org/10.1145/3394171.3413678 |
| [36] |
A. Zadeh, P. P. Liang, N. Mazumder, S. Poria, E. Cambria, L. P. Morency, Memory fusion network for multi-view sequential learning, AAAI Press, (2018), 5634–5641. https://doi.org/10.1609/aaai.v32i1.12021 doi: 10.1609/aaai.v32i1.12021
|
| [37] |
S. Siriwardhana, T. Kaluarachchi, M. Billinghurst, S. Nanayakkara, Multimodal emotion recognition with transformer-based self supervised feature fusion, IEEE Access, 8 (2020), 176274–176285. https://doi.org/10.1109/ACCESS.2020.3026823 doi: 10.1109/ACCESS.2020.3026823
|
| [38] |
K. Kim, S. Park, AOBERT: All-modalities-in-One BERT for multimodal sentiment analysis, Inf. Fusion, 92 (2023), 37–45. https://doi.org/10.1016/j.inffus.2022.11.022 doi: 10.1016/j.inffus.2022.11.022
|
| [39] | W. Han, H. Chen, S Poria, Improving multimodal fusion with hierarchical mutual information maximization for multimodal sentiment analysis, in Proceedings of the 2021 Conference on Empirical Methods in Natural Language Processing, Association for Computational Linguistics, (2021), 9180–9192. https://doi.org/10.18653/v1/2021.emnlp-main.723 |
| [40] |
S. Mai, Y. Zeng, S. Zheng, H. Hu, Hybrid contrastive learning of tri-modal representation for multimodal sentiment analysis, IEEE Trans. Affective Comput., 14 (2023), 2276–2289. https://doi.org/10.1109/TAFFC.2022.3172360 doi: 10.1109/TAFFC.2022.3172360
|












Figures(13) / Tables(7)

Zhijing Xu, Yang Gao. Research on cross-modal emotion recognition based on multi-layer semantic fusion[J]. Mathematical Biosciences and Engineering, 2024, 21(2): 2488-2514. doi: 10.3934/mbe.2024110







 DownLoad:
DownLoad: