Citation: Zhenquan Zhang, Junhao Liang, Zihao Wang, Jiajun Zhang, Tianshou Zhou. Modeling stochastic gene expression: From Markov to non-Markov models[J]. Mathematical Biosciences and Engineering, 2020, 17(5): 5304-5325. doi: 10.3934/mbe.2020287

| [1] | A. Sanchez, S. Choubey, J. Kondev, Stochastic models of transcription: From single molecules to single cells, Methods, 62 (2013), 13-25. |
| [2] | J. Zhang, T. Zhou, Promoter-mediated transcriptional dynamics, Biophys. J., 106 (2014), 479-488. |
| [3] | T. Zhou, J. Zhang, Analytical results for a multistate gene model, SIAM J. Appl. Math., 72 (2012), 789-818. |
| [4] | L. Cai, N. Friedman, X. S. Xie, Stochastic protein expression in individual cells at the single molecule level, Nature, 440 (2006), 358-362. |
| [5] | T. Liu, J. Zhang, T. Zhou, Effect of interaction between chromatin loops on cell-to-cell variability in gene expression, PLoS Comput. Biol., 12 (2016), e1004917. |
| [6] | D. R. Rigney, W. C. Schieve, Stochastic model of linear, continuous protein-synthesis in bacterial populations, J. Theor. Biol., 69 (1977), 761-766. |
| [7] | O. G. Berg, A model for the statistical fluctuations of protein numbers in a microbial population, J. Theor. Biol., 71 (1978), 587-603. |
| [8] | P. K. Tapaswi, R. K. Roychoudhury, T. Prasad, A stochastic model of gene activation and RNA synthesis during embryogenesis, Sankhyā Indian J. Statist. Ser. B, 49 (1987), 51-67. |
| [9] | J. Peccoud, B. Ycart, Markovian modeling of gene-product synthesis, Theor. Popul. Biol., 48 (1995), 222-234. |
| [10] | D. R. Rigney, Stochastic model of constitutive protein levels in growing and dividing bacterial cells, J. Theor. Biol., 76 (4), 453-480. |
| [11] | D. R. Rigney, Stochastic models of cellular variability. In Kinetic Logic A Boolean Approach to the Analysis of Complex Regulatory Systems, Springer, Berlin, Heidelberg, 1979. |
| [12] | T. B. Kepler, T. C. Elston, Stochasticity in transcriptional regulation: origins, consequences, and mathematical representations, Biophys. J., 81 (2001), 3116-3136. |
| [13] | M. Thattai, A. Van Oudenaarden, Intrinsic noise in gene regulatory networks, Proc. Natl. Acad. Sci. U.S.A., 98 (2001), 8614-8619. |
| [14] | P. S. Swain, M. B. Elowitz, E. D. Siggia, Intrinsic and extrinsic contributions to stochasticity in gene expression, Proc. Natl. Acad. Sci. U.S.A., 99 (2002), 12795-12800. |
| [15] | M. Sasai, P. G. Wolynes, Stochastic gene expression as a many-body problem, Proc. Natl. Acad. Sci. U.S.A., 100 (2003), 2374-2379. |
| [16] | T. Jia, R. V. Kulkarni, Intrinsic noise in stochastic models of gene expression with molecular memory and bursting, Phys. Rev. Lett., 106 (2011), 058102. |
| [17] | Z. Cao, R. Grima, Linear mapping approximation of gene regulatory networks with stochastic dynamics, Nat. Commun., 9 (2018), 1-15. |
| [18] | C. V. Harper, B. Finkenstädt, D. J. Woodcock, S. Friedrichsen, S. Semprini, L. Ashall, et al., Dynamic analysis of stochastic transcription cycles, PLoS Biol., 9 (2011), e1000607. |
| [19] | M. R. Green, Eukaryotic transcription activation: Right on target, Mol. Cell, 18 (2005), 399-402. |
| [20] | J. Paulsson, Models of stochastic gene expression, Phys. Life Rev., 2 (2005), 157-175. |
| [21] | G. Hornung, R. Bar-Ziv, D. Rosin, N. Tokuriki, D. S. Tawfik, M. Oren, et al., Noise-mean relationship in mutated promoters, Genom. Res., 22 (2012), 2409-2417. |
| [22] | Q. Li, G. Barkess, H. Qian, Chromatin looping and the probability of transcription, Trend. Genet., 22 (2006), 197-202. |
| [23] | C. W. Gardiner, Handbook of Stochastic Methods for Physics, Chemistry and the Natural Sciences, Springer, Berlin, Heidelberg, 2004. |
| [24] | J. D. Jordan, E. M. Landau, R. Iyengar, Signaling networks: The origins of cellular multitasking, Cell, 103 (2000), 193-200. |
| [25] | L. Bintu, J. Yong, Y. E. Antebi, K. McCue, Y. Kazuki, N. Uno, et al., Dynamics of epigenetic regulation at the single-cell level, Science, 351 (2016), 720-724. |
| [26] | C. W. Gardiner, Stochastic Methods: a handbook for the natural and social sciences, Springer, New York, 2009. |
| [27] | N. G. Van Kampen, Stochastic Processes in Physics and Chemistry, North-Holland, Amsterdam, 2007. |
| [28] | E. Pardoux, Markov Processes and Applications: Algorithms, Networks, Genome and Finance, vol 796, John Wiley & Sons, New York, 2008. |
| [29] | H. Andersson, T. Britton, Stochastic epidemic models and their statistical analysis, vol. 151, Springer Science & Business Media, 2012. |
| [30] | M. Salathé, M. Kazandjieva, J. W. Lee, P. Levis, M. W. Feldman, J. H. Jones, A high-resolution human contact network for infectious disease transmission, Proc. Natl. Acad. Sci. U.S.A., 107 (2010), 22020-22025. |
| [31] | A. Corral, Long-term clustering, scaling, and universality in the temporal occurrence of earthquakes, Phys. Rev. Lett., 92 (2004), 108501. |
| [32] | P. S. Stumpf, R. C. Smith, M. Lenz, A. Schuppert, F. J. Müller, A. Babtie, et al., Stem cell differentiation as a non-Markov stochastic process, Cell Syst., 5 (2017), 268-282. |
| [33] | D. M. Suter, N. Molina, D. Gatfield, K. Schneider, U. Schibler, F. Naef, Mammalian genes are transcribed with widely different bursting kinetics, Science, 332 (2011), 472-474. |
| [34] | T. Guérin, O. Bénichou, R. Voituriez, Non-Markovian polymer reaction kinetics, Nat. Chem., 4 (2012), 568-573. |
| [35] | A. L. Barabasi, The origin of bursts and heavy tails in human dynamics, Nature, 435 (2005), 207-211. |
| [36] | J. M. Pedraza, J. Paulsson, Effects of molecular memory and bursting on fluctuations in gene expression, Science, 319 (2008), 339-343. |
| [37] | G. Srinivasan, D. M. Tartakovsky, B. A. Robinson, A. B. Aceves, Quantification of uncertainty in geochemical reactions, Water Resour. Res., 43 (2007), W12415. |
| [38] | S. Condamin, O. Bénichou, V. Tejedor, R. Voituriez, J. Klafter, First-passage times in complex scale-invariant media, Nature, 450 (2007), 77-80. |
| [39] | G. Guigas, M. Weiss, Sampling the cell with anomalous diffusion—the discovery of slowness, Biophys. J., 94 (2008), 90-94. |
| [40] | Y. Meroz, I. M. Sokolov, J. Klafter, Distribution of first-passage times to specific targets on compactly explored fractal structures, Phys. Rev. E, 83 (2011), 020104. |
| [41] | M. Dentz, A. Russian, P. Gouze, Self-averaging and ergodicity of subdiffusion in quenched random media, Phys. Rev. E, 93 (2016), 010101. |
| [42] | A. A. Ovchinnikov, Y. B. Zeldovich, Role of density fluctuations in bimolecular reaction kinetics, Chem. Phys., 28 (1978), 215-218. |
| [43] | M. Dobrzyński, F. J. Bruggeman, Elongation dynamics shape bursty transcription and translation, Proc. Natl. Acad. Sci. U.S.A., 106 (2009), 2583-2588. |
| [44] | D. R. Larson, D. Zenklusen, B. Wu, J. A. Chao, R. H. Singer, Real-time observation of transcription initiation and elongation on an endogenous yeast gene, Science, 332 (2011), 475-478. |
| [45] | S. Yunger, L. Rosenfeld, Y. Garini, Y. Shav-Tal, Single-allele analysis of transcription kinetics in living mammalian cells, Nat. Methods, 7 (2010), 631-633. |
| [46] | I. Golding, J. Paulsson, S. M. Zawilski, E. C. Cox, Real-time kinetics of gene activity in individual bacteria, Cell, 123 (2005), 1025-1036. |
| [47] | T. Muramoto, D. Cannon, M. Gierliński, A. Corrigan, G. J. Barton, J. R. Chubb, Live imaging of nascent RNA dynamics reveals distinct types of transcriptional pulse regulation, Proc. Natl. Acad. Sci. U.S.A., 109 (2012), 7350-7355. |
| [48] | A. Raj, C. S. Peskin, D. Tranchina, D. Y. Vargas, S. Tyagi, Stochastic mRNA synthesis in mammalian cells, PLoS Biol., 4 (2006), e309. |
| [49] | D. G. Spiller, C. D. Wood, D. A. Rand, M. R. White, Measurement of single-cell dynamics, Nature, 465 (2010), 736-745. |
| [50] | A. Eldar, M. B. Elowitz, Functional roles for noise in genetic circuits, Nature, 467 (2010), 167-173. |
| [51] | B. Zoller, D. Nicolas, N. Molina, F. Naef, Structure of silent transcription intervals and noise characteristics of mammalian genes, Mol. Syst. Biol., 11 (2015), 823. |
| [52] | T. R. Sokolowski, T. Erdmann, P. R. Ten Wolde, Mutual repression enhances the steepness and precision of gene expression boundaries, PLoS Comput. Biol., 8 (2012), e1002654. |
| [53] | J. Paulsson, Summing up the noise in gene networks, Nature, 427 (2001), 415-418. |
| [54] | D. R. Larson, What do expression dynamics tell us about the mechanism of transcription?, Curr. Opin. Gen. Dev., 21 (2011), 591-599. |
| [55] | V. Shahrezaei, P. S. Swain, Analytical distributions for stochastic gene expression, Proc. Natl. Acad. Sci. U.S.A., 105 (2008), 17256-17261. |
| [56] | N. Kumar, A. Singh, R. V. Kulkarni, Transcriptional bursting in gene expression: analytical results for general stochastic models, PLoS Comput. Biol., 11 (2015), e1004292. |
| [57] | Z. Wang, Z. Zhang, T. Zhou, Exact distributions for stochastic models of gene expression with arbitrary regulation, Sci. China Math., 63 (2020), 485-500. |
| [58] | P. Liu, Z. Yuan, L. Huang, T. Zhou, Roles of factorial noise in inducing bimodal gene expression, Phys. Rev. E, 91 (2015), 062706. |
| [59] | J. Zhang, Q. Nie, T. Zhou, A moment-convergence method for stochastic analysis of biochemical reaction networks, J. Chem. Phys., 144 (2016), 194109. |
| [60] | A. B. O. Daalhuis, Confluent hypergeometric functions, NIST Handb. Math. Funct., 2010. |
| [61] | T. Aquino, M. Dentz, Chemical continuous time random walks, Phys. Rev. Lett., 119 (2017), 230601. |
| [62] | N. Masuda, M. A. Porter, R. Lambiotte, Random walks and diffusion on networks, Phys. Rep., 716 (2017), 1-58. |
| [63] | R. Kutner, J. Masoliver, The continuous time random walk, still trendy: fifty-year history, state of art and outlook, Eur. Phys. J. B, 90 (2017), 50. |
| [64] | L. Liu, B. R. K. Kashyap, J. G. C. Templeton, On the GIX/G/∞ system, J. Appl. Prob., 27 (1990), 671-683. |
| [65] | A. R. Stinchcombe, C. S. Peskin, D. Tranchina, Population density approach for discrete mRNA distributions in generalized switching models for stochastic gene expression, Phys. Rev. E, 85 (2012), 061919. |
| [66] | N. Masuda, L. E. Rocha, A Gillespie algorithm for non-Markovian stochastic processes, SIAM Rev., 60 (2018), 95-115. |
| [67] | C. Deneke, R. Lipowsky, A. Valleriani, Complex degradation processes lead to non-exponential decay patterns and age-dependent decay rates of messenger RNA, PloS One, 8 (2013), e55442. |
| [68] | B. C. Arnold, Majorization: Here, there and everywhere, Statist. Sci., 22 (2007), 407-413. |
| [69] | A. David, S. Larry, The least variable phase type distribution is Erlang, Stochastic Models, 3 (1987), 467-473. |
| [70] | J. Zhang, T. Zhou, Markovian approaches to modeling intracellular reaction processes with molecular memory, Proc. Natl. Acad. Sci. U.S.A., 116 (2019), 23542-23550. |
| [71] | H. Qiu, B. Zhang, T. Zhou, Analytical results for a generalized model of bursty gene expression with molecular memory, Phys. Rev. E, 100 (2019), 012128. |
| [72] | A. Coulon, C. C. Chow, R. H. Singer, D. R. Larson, Eukaryotic transcriptional dynamics: From single molecules to cell populations, Nat. Rev. Genet., 14 (2013), 572-584. |
| [73] | W. J. Blake, M. Kærn, C. R. Cantor, J. J. Collins, Noise in eukaryotic gene expression, Nature, 422 (2003), 633-637. |
| [74] | J. M. Raser, E. K. O'Shea, Control of stochasticity in eukaryotic gene expression, Science, 304 (2004), 1811-1814. |
| [75] | N. Friedman, L. Cai, X. S. Xie, Linking stochastic dynamics to population distribution: an analytical framework of gene expression, Phys. Rev. Lett., 97 (2006), 168302. |
| [76] | A. M. Kringstein, F. M. Rossi, A. Hofmann, H. M. Blau, Graded transcriptional response to different concentrations of a single transactivator, Proc. Natl. Acad. Sci. U.S.A., 95 (1998), 13670-13675. |
| [77] | J. Stewart-Ornstein, C. Nelson, J. DeRisi, J. S. Weissman, H. El-Samad, Msn2 coordinates a stoichiometric gene expression program, Curr. Biol., 23 (2013), 2336-2345. |
| [78] | J. Paulsson, M. Ehrenberg, Noise in a minimal regulatory network: plasmid copy number control, Quart. Rev. Biophys., 34 (2001), 1-59. |
| [79] | M. B. Elowitz, A. J. Levine, E. D. Siggia, P. S. Swain, Stochastic gene expression in a single cell, Science, 297 (2002), 1183-1186. |
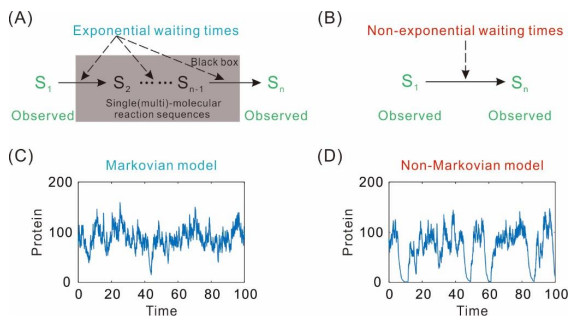
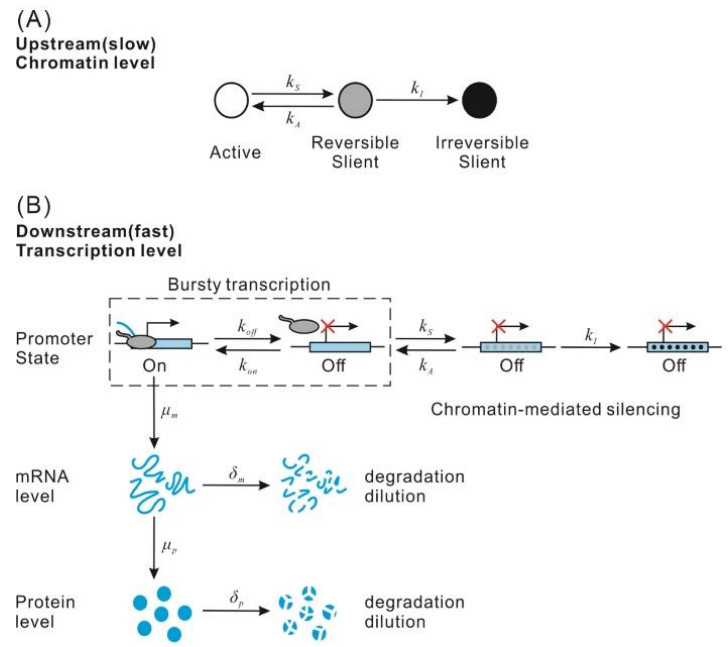
Figures(2)

Zhenquan Zhang, Junhao Liang, Zihao Wang, Jiajun Zhang, Tianshou Zhou. Modeling stochastic gene expression: From Markov to non-Markov models[J]. Mathematical Biosciences and Engineering, 2020, 17(5): 5304-5325. doi: 10.3934/mbe.2020287







 DownLoad:
DownLoad: