Citation: Harish Garg, Nancy. Algorithms for single-valued neutrosophic decision making based on TOPSIS and clustering methods with new distance measure[J]. AIMS Mathematics, 2020, 5(3): 2671-2693. doi: 10.3934/math.2020173

| [1] |
L. A. Zadeh, Fuzzy sets, Information and Control, 8 (1965), 338-353. doi: 10.1016/S0019-9958(65)90241-X

|
| [2] |
K. T. Atanassov, Intuitionistic fuzzy sets, Fuzzy Set. Syst., 20 (1986), 87-96. doi: 10.1016/S0165-0114(86)80034-3
|
| [3] | F. Smarandache, Neutrosophy. Neutrosophic Probability, Set, and Logic, ProQuest Information & Learning, Ann Arbor, Michigan, USA, 1998. |
| [4] | H. Wang, F. Smarandache, Y. Q. Zhang, et al. Single valued neutrosophic sets, Multispace Multistructure, 4 (2010), 410-413. |
| [5] |
J. Ye, A multicriteria decision-making method using aggregation operators for simplified neutrosophic sets, J. Intell. Fuzzy Syst., 26 (2014), 2459-2466. doi: 10.3233/IFS-130916
|
| [6] |
J. J. Peng, J. Q. Wang, J. Wang, et al. Simplified neutrosophic sets and their applications in multicriteria group decision-making problems, International Journal of System Science, 47 (2016), 2342-2358. doi: 10.1080/00207721.2014.994050
|
| [7] |
Nancy, H. Garg, An improved score function for ranking neutrosophic sets and its application to decision-making process, International Journal for Uncertainty Quantification, 6 (2016), 377-385. doi: 10.1615/Int.J.UncertaintyQuantification.2016018441
|
| [8] |
D. Rani, H. Garg, Some modified results of the subtraction and division operations on interval neutrosophic sets, J. Exp. Theor. Artif. In., 31 (2019), 677-698. doi: 10.1080/0952813X.2019.1592236
|
| [9] | P. Liu, Y. Chu, Y. Li, et al. Some generalized neutrosophic number hamacher aggregation operators and their application to group decision making, Int. J. Fuzzy Syst., 16 (2014), 242-255. |
| [10] |
Nancy, H. Garg, Novel single-valued neutrosophic decision making operators under Frank norm operations and its application, Int. J. Uncertain. Quan., 6 (2016), 361-375. doi: 10.1615/Int.J.UncertaintyQuantification.2016018603
|
| [11] |
H. Garg, Nancy, New logarithmic operational laws and their applications to multiattribute decision making for single-valued neutrosophic numbers, Cogn. Syst. Res., 52 (2018), 931-946. doi: 10.1016/j.cogsys.2018.09.001
|
| [12] |
G. Wei, Z. Zhang, Some single-valued neutrosophic Bonferroni power aggregation operators in multiple attribute decision making, Journal of Ambient Intelligence and Humanized Computing, 10 (2019), 863-882. doi: 10.1007/s12652-018-0738-y
|
| [13] | L. Yang, B. Li, A multi-criteria decision-making method using power aggregation operators for single-valued neutrosophic sets, International Journal of Database and Theory and Application, 9 (2016), 23-32. |
| [14] | H. Garg, Nancy, Linguistic single-valued neutrosophic power aggregation operators and their applications to group decision-making problems, IEEE/CAA Journal of Automatic Sinica, 7 (2020), 546-558. |
| [15] |
P. Ji, J. Q. Wang, H. Y. Zhang, Frank prioritized Bonferroni mean operator with single-valued neutrosophic sets and its application in selecting third-party logistics providers, Neural Comput. Appl., 30 (2018), 799-823. doi: 10.1007/s00521-016-2660-6
|
| [16] | H. Garg, Novel neutrality aggregation operators-based multiattribute group decision making method for single-valued neutrosophic numbers, Soft Comput., 2019, 1-23. |
| [17] | H. Garg, Nancy, Multiple criteria decision making based on frank choquet heronian mean operator for single-valued neutrosophic sets, Applied and Computational Mathematics, 18, (2019), 163-188. |
| [18] | P. Majumdar, Neutrosophic Sets and Its Applications to Decision Making, Computational Intelligence for Big Data Analysis, Springer, Cham, 2015. |
| [19] |
H. L. Huang, New distance measure of single-valued neutrosophic sets and its application, Int. J. Intell. Syst., 31 (2016), 1021-1032. doi: 10.1002/int.21815
|
| [20] | C. Liu, Y. Luo, The weighted distance measure based method to neutrosophic multiattribute group decision making, Math. Probl. Eng., 2016 (2016), 3145341. |
| [21] | H. Garg, Nancy, Some new biparametric distance measures on single-valued neutrosophic sets with applications to pattern recognition and medical diagnosis, Information 8 (2017), 162. |
| [22] |
X. H. Wu, J. Q. Wang, J. J. Peng, et al. Cross - entropy and prioritized aggregation operator with simplified neutrosophic sets and their application in multi-criteria decision-making problems, International Journal of Fuzzy Systems, 18 (2016), 1104-1116. doi: 10.1007/s40815-016-0180-2
|
| [23] | H. Garg, Nancy, On single-valued neutrosophic entropy of order α, Neutrosophic Sets and Systems, 14 (2016), 21-28. |
| [24] | K. Mondal, S. Pramanik, Neutrosophic tangent similarity measure and its application to multiple attribute decision making, Neutrosophic Sets and Systems, 9 (2015), 80-87. |
| [25] | K. Mondal, S. Pramanik, B. C. Giri, Hybrid binary logarithm similarity measure for MAGDM problems under SVNS assessments, Neutrosophic Sets and Systems, 20 (2018), 12-25. |
| [26] |
F. Liu, G. Aiwu, V. Lukovac, et al. A multicriteria model for the selection of the transport service provider: A single valued neutrosophic DEMATEL multicriteria model, Decision Making: Applications in Management and Engineering, 1 (2018), 121-130. doi: 10.31181/dmame1801121r
|
| [27] |
Nancy, H. Garg, A novel divergence measure and its based TOPSIS method for multi criteria decision - making under single - valued neutrosophic environment, J. Intell. Fuzzy Syst., 36 (2019), 101-115. doi: 10.3233/JIFS-18040
|
| [28] | C. L. Hwang, K. Yoon, Multiple Attribute Decision Making Methods and Applications A State-ofthe-Art Survey, Springer-Verlag Berlin Heidelberg, 1981. |
| [29] |
P. Biswas, S. Pramanik, B. C. Giri, TOPSIS method for multi-attribute group decision-making under single-valued neutrosophic environment, Neural Computing and Applications, 27 (2016), 727-737. doi: 10.1007/s00521-015-1891-2
|
| [30] |
H. Pouresmaeil, E. Shivanian, E. Khorram, et al. An extended method using TOPSIS and VIKOR for multiple attribute decision making with multiple decision makers and single valued neutrosophic numbers, Advances and Applications in Statistics, 50 (2017), 261-292. doi: 10.17654/AS050040261
|
| [31] | G. Selvachandran, S. Quek, F. Smarandache, et al. An extended technique for order preference by similarity to an ideal solution (TOPSIS) with maximizing deviation method based on integrated weight measure for single-valued neutrosophic sets, Symmetry, 10 (2018), 236. |
| [32] |
X. D. Peng, J. G. Dai, Approaches to single-valued neutrosophic MADM based on MABAC, TOPSIS and new similarity measure with score function, Neural Computing and Applications, 29 (2018), 939-954. doi: 10.1007/s00521-016-2607-y
|
| [33] |
I. Mukhametzyanov, D. Pamucar, A sensitivity analysis in MCDM problems: A statistical approach, Decision making: Applications in Management and Engineering, 1 (2018), 51-80. doi: 10.31181/dmame180151b
|
| [34] |
E. H. Ruspini, A new approach to clustering, Information and control, 15 (1969), 22-32. doi: 10.1016/S0019-9958(69)90591-9
|
| [35] | J. Ye, Single-valued neutrosophic minimum spanning tree and its clustering method, Journal of intelligent Systems, 23 (2014), 311-324. |
| [36] | J. Ye, Clustering methods using distance-based similarity measures of single-valued neutrosophic sets, Journal of Intelligent Systems, 23 (2014), 379-389. |
| [37] |
Y. Guo, A. Şengür, A novel image segmentation algorithm based on neutrosophic similarity clustering, Applied Soft Computing, 25 (2014), 391-398. doi: 10.1016/j.asoc.2014.08.066
|
| [38] | J. Ye, A netting method for clustering-simplified neutrosophic information, Soft Computing, 21 (2016), 7571-7577. |
| [39] |
N. D. Thanh, M. Ali, L. H. Son, A novel clustering algorithm in a neutrosophic recommender system for medical diagnosis, Cognitive Computation, 9 (2017), 526-544. doi: 10.1007/s12559-017-9462-8
|
| [40] |
A. S. Ashour, Y. Guo, E. Kucukkulahli, et ai. A hybrid dermoscopy images segmentation approach based on neutrosophic clustering and histogram estimation, Applied Soft Computing, 69 (2018), 426-434. doi: 10.1016/j.asoc.2018.05.003
|
| [41] |
X. Wang, E. Triantaphyllou, Ranking irregularities when evaluating alternatives by using some ELECTRE methods, Omega - International Journal of Management Science, 36 (2008), 45-63. doi: 10.1016/j.omega.2005.12.003
|
| [42] |
Z. S. Xu, J. Chen, J. J. Wu, Cluster algorithm for intuitionistic fuzzy sets, Information Sciences, 178 (2008), 3775-3790. doi: 10.1016/j.ins.2008.06.008
|
| [43] |
M. Noureddine, M. Ristic, Route planning for hazardous materials transportation: Multicriteria decision making approach, Decision Making: Applications in Management and Engineering, 2 (2019), 66-85. doi: 10.31181/dmame1901066n
|
| [44] | H. Garg, G. Kaur, Quantifying gesture information in brain hemorrhage patients using probabilistic dual hesitant fuzzy sets with unknown probability information, Computers and Industrial Engineering, 140 (2020), 106211. |
| [45] | H. Garg, G. Kaur, A robust correlation coefficient for probabilistic dual hesitant fuzzy sets and its applications, Neural Computing & Applications, 2019 (2019), 1-20. |
| [46] |
H. Garg, Nancy, Algorithms for possibility linguistic single-valued neutrosophic decision-making based on COPRAS and aggregation operators with new information measures, Measurement, 138 (2019), 278-290. doi: 10.1016/j.measurement.2019.02.031
|
| [47] |
H. Garg, Nancy, Non-linear programming method for multi-criteria decision making problems under interval neutrosophic set environment, Applied Intelligence, 48 (2018), 2199-2213. doi: 10.1007/s10489-017-1070-5
|
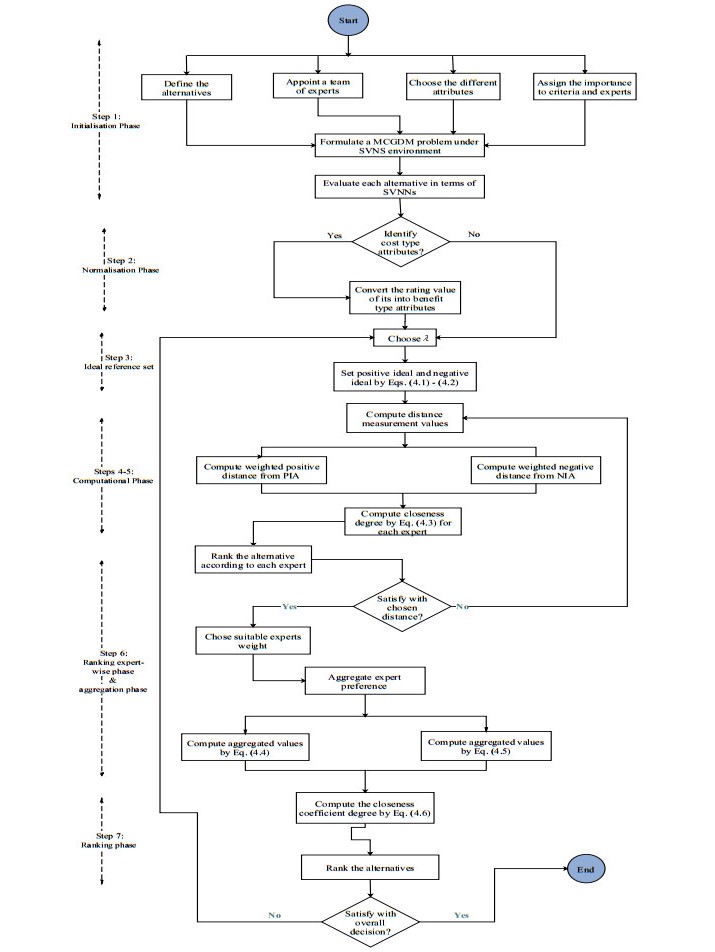
Figures(1) / Tables(8)

Harish Garg, Nancy. Algorithms for single-valued neutrosophic decision making based on TOPSIS and clustering methods with new distance measure[J]. AIMS Mathematics, 2020, 5(3): 2671-2693. doi: 10.3934/math.2020173







 DownLoad:
DownLoad: