Citation: Alireza Moravveji, Mansour Sayyah, Elham Shamsnia, Zarichehr Vakili. Comparing the prolonged effect of interval versus continuous aerobic exercise on blood inflammatory marker of Visfatin level and body mass index of sedentary overweigh/fat female college students[J]. AIMS Public Health, 2019, 6(4): 568-576. doi: 10.3934/publichealth.2019.4.568

| [1] |
Poirier P, Giles TD, Bray GA, et al. (2006) Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 113: 898–918. doi: 10.1161/CIRCULATIONAHA.106.171016

|
| [2] |
Zalesin KC, Franklin BA, Miller WM, et al. (2011) Impact of obesity on cardiovascular disease. Med Clin North Am 95: 919–937. doi: 10.1016/j.mcna.2011.06.005
|
| [3] |
Pandey A, Berry JD, Lavie CJ (2015) Cardiometabolic disease leading to heart failure: better fat and fit than lean and lazy. Curr Heart Fail Rep 12: 302–308. doi: 10.1007/s11897-015-0265-5
|
| [4] |
Aune D, Sen A, Norat T, et al. (2016) Body mass index, abdominal fatness, and heart failure incidence and mortality: a systematic review and dose-response meta-analysis of prospective studies. Circulation 133: 639–649. doi: 10.1161/CIRCULATIONAHA.115.016801
|
| [5] | Ndumele CE, Matsushita K, Lazo M, et al. (2016) Obesity and Subtypes of Incident Cardiovascular Disease. J Am Heart Assoc DOI: 10.1161/JAHA.116.003921 |
| [6] |
Yatsuya H, Yamagishi K, North KE, et al. (2010) Associations of obesity measures with subtypes of ischemic stroke in the ARIC Study. J Epidemiol 20: 347–354. doi: 10.2188/jea.JE20090186
|
| [7] | Lu Y, Hajifathalian K, Ezzati M, et al. (2014) Metabolic mediators of the effects of body‐mass index, overweight, and obesity on coronary heart disease and stroke: a pooled analysis of 97 prospective cohorts with 1.8 million participants. Lancet 383: 970–983. |
| [8] |
Morkedal B, Vatten LJ, Romundstad PR, et al. (2014) Risk of myocardial infarction and heart failure among metabolically healthy but obese individuals: HUNT [Nord‐Trondelag Health Study], Norway. J Am Coll Cardiol 63:1071–1078. doi: 10.1016/j.jacc.2013.11.035
|
| [9] |
Guh DP, Zhang W, Bansback N, et al. (2009) The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health 9: 88. doi: 10.1186/1471-2458-9-88 doi: 10.1186/1471-2458-9-88
|
| [10] |
Everson SA, Goldberg DE, Helmrich SP, et al. (1998) Weight gain and the risk of developing insulin resistance syndrome. Diabetes Care 21: 1637–1643. doi: 10.2337/diacare.21.10.1637
|
| [11] |
Abdullah A, Peeters A, de Courten M, et al. (2010) The magnitude of association between overweight and obesity and the risk of diabetes: a meta-analysis of prospective cohort studies. Diabetes Res Clin Pract 89: 309–319. doi: 10.1016/j.diabres.2010.04.012 doi: 10.1016/j.diabres.2010.04.012
|
| [12] | Dudina A, Cooney MT, Bacquer DD, et al. (2011) SCORE Investigators. Relationships between body mass index, cardiovascular mortality, and risk factors: a report from the SCORE investigators. Eur J Cardiovasc Prev Rehabil 18: 731–742. doi: 10.1177/1741826711412039 |
| [13] | Nejmeddine O, Marwa K, Sami B, et al. (2014) Effects of a high-intensity interval training program on aerobic capacity and lipid profile in trained subjects. J Sports Med 5: 243–248. doi: 10.2147/OAJSM.S68701PMCID: PMC4207574 |
| [14] | Bouassida A, Chamari K, Zaouali M, et al. (2008) Review on leptin and adiponectin responses and adaptations to acute and chronic exercise. Br J Sports Med 44: 620–630. |
| [15] |
Hayashino Y, Jackson JL, Hirata T, et al. (2014) Effects of exercise on C-reactive protein, inflammatory cytokine and adipokine in patients with type 2 diabetes: a meta-analysis of randomized controlled trials. Metabolism 63: 431–440. doi: 10.1016/j.metabol.2013.08.018
|
| [16] |
Jamurtas AZ, Stavropoulos-Kalinoglou A, Koutsias S, et al. (2015) Adiponectin, Resistin, and Visfatin in Childhood Obesity and Exercise. Pediatr Exerc Sci 27: 454–462. doi: 10.1123/pes.2014-0072
|
| [17] | Raygan F, Sayyah M, Janesar Qamsari SMR, et al. (2017) Effects of Submaximal Aerobic Exercise on Regulatory T Cell Markers of Male Patients Suffering from Ischemic Heart Disease. Iran J Allergy Asthma Immunol 16: 14–20. |
| [18] | Ahmadizadeh R, Bassami M, Tahmasbi V (2012) Relationship rest level of visfatin and its change in response of acute endurance training with aerobic fitness and body composition in healthy men. Sport Physiplogy 15: 27–38. |
| [19] |
Vadeboncoeur C, Foster C, Townsend N (2016) Freshman 15 in England: a longitudinal evaluation of first year university student's weight change. BMC Obesity 3: 45 DOI: 10.1186/s40608-016-0125-1 doi: 10.1186/s40608-016-0125-1
|
| [20] |
Fukuhara A, Matsuda M, Nishizawa M, et al. (2005) Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science 307: 426 – 430. doi: 10.1126/science.1097243
|
| [21] |
Berndt J, Kl Ö ting N, Kralisch S, et al. (2005) Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes 54: 2911–2916. doi: 10.2337/diabetes.54.10.2911
|
| [22] | Taghian F, Zolfaghary M, Hedayati M (2014) Effect of 12 weeks aerobic exercise on visfatin level and insulin resistance in obese women. Razi J Med Sci 20: 35–44. |
| [23] | Fantuzzi G, Faggioni R (2000) Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol 68: 437–446. |
| [24] |
Irving AJ, Wallace L, Durakoglugil D, et al. (2006) Leptin enhances NR2B-mediated N-methyl-D-aspartate responses via a mitogenactivated protein kinase-dependent process in cerebellar granule cells. Neuroscience 138: 1137–1148. doi: 10.1016/j.neuroscience.2005.11.042
|
| [25] |
Fukuhara A, Matsuda M, Nishizawa M, et al. (2005) Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science 307: 426–430. doi: 10.1126/science.1097243
|
| [26] |
Choi KM, Kim JH, Cho GJ, et al. (2007) Effect of exercise training on plasma visfatin and eotaxin levels. Eur J Endocrinol 157: 437–442. doi: 10.1530/EJE-07-0127
|
| [27] |
Haider DG, Mittermayer F, Schaller G, et al. (2006) Free fatty acids normalize arosiglitazone-induced visfatin release. Am J Physiol Endocrinol Metab 291: E885–890. doi: 10.1152/ajpendo.00109.2006
|
| [28] |
Brema I, Hatunic M, Finucane F, et al. (2008) Plasma visfatin is reduced after aerobic exercise in early onset type 2 diabetes mellitus. Diabetes Obes Metab 10: 600–602. doi: 10.1111/j.1463-1326.2008.00872.x
|
| [29] |
Gao Y, Wang C, Pan T, et al. (2014) Impact of metformin treatment and swimming exercise on visfatin levels in high-fat-induced obesity rats. Arq Bras Endocrinol Metab 58: 42–47. doi: 10.1590/0004-2730000002840
|
| [30] | Seifi L, Daryanoosh F, Samadi M (2016) The effect of 12 weeks aerobic exercise training on visfatin, chemerin serum changes in 45–60 years old obese women with diabetes type 2. J Shahid Sadoughi Univ Med Sci 24: 55–64. |
| [31] |
Finkelstein EA, Khavjou OA, Thompson H, et al. (2012) Obesity and severe obesity forecasts through 2030. Am J Prev Med 42: 563–570. doi: 10.1016/j.amepre.2011.10.026
|
| [32] |
Sayyah M, Rahimi SM, Bigdeli M, et al. (2011) Comparing the anthropometric characteristics of injured and non-injured girl student athletes participating in the sport olympiads held by the ministry of health and medical education in the summer of 2009 in the city of Yazd. Biosci Biotechnol Res Asia 8: 367–371. doi: 10.13005/bbra/875
|
| [33] | Salehi A, Ehtram H, SaeidSoukhtehzari MS, et al. (2013) Effects of two months of physical activity on the copper level of overweight sedentary young male and female measured at nano scale level. Life Sci J 3: 10. |
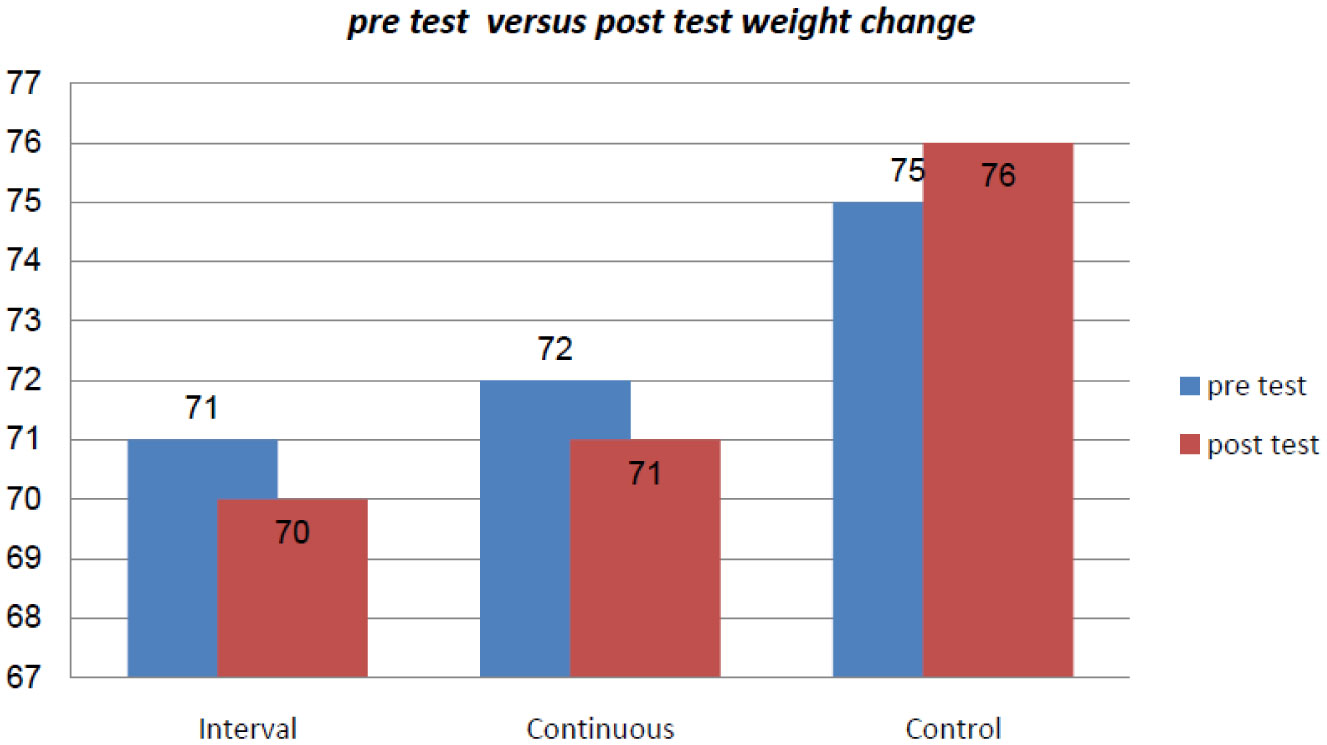
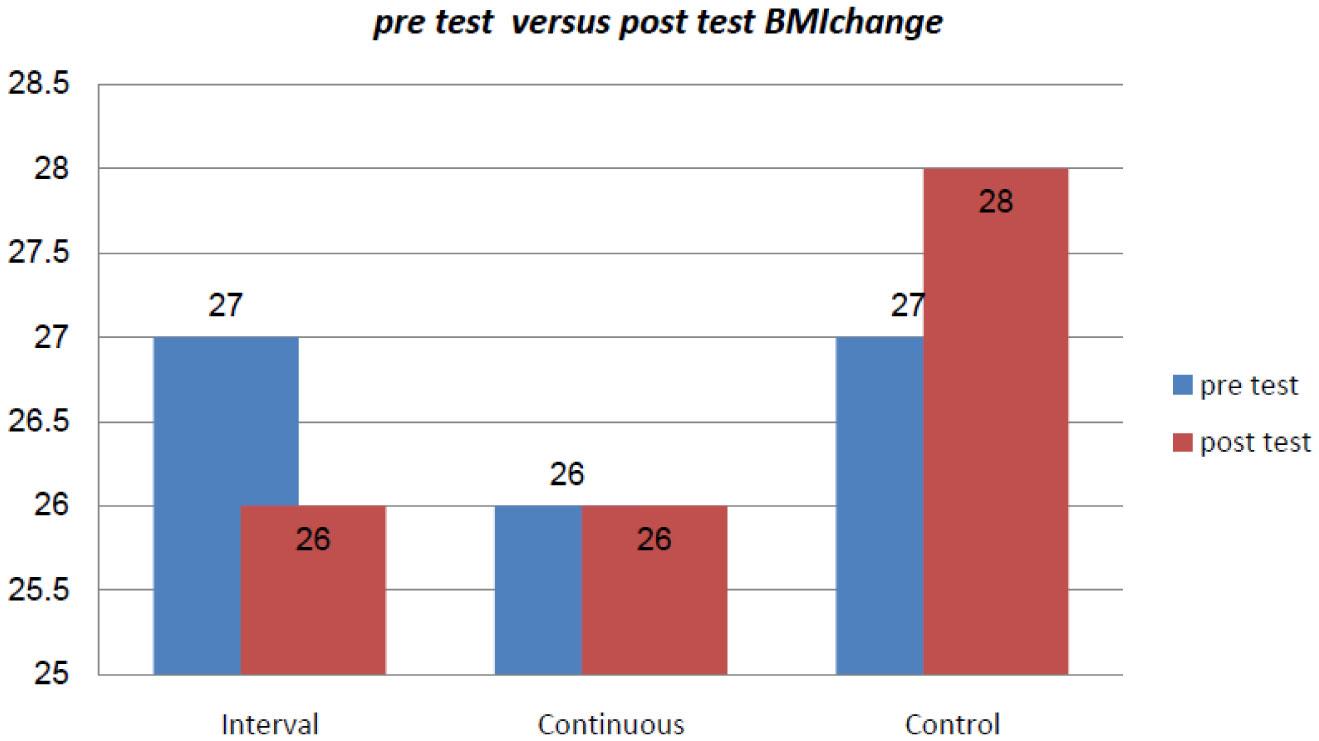
Figures(2) / Tables(2)

Alireza Moravveji, Mansour Sayyah, Elham Shamsnia, Zarichehr Vakili. Comparing the prolonged effect of interval versus continuous aerobic exercise on blood inflammatory marker of Visfatin level and body mass index of sedentary overweigh/fat female college students[J]. AIMS Public Health, 2019, 6(4): 568-576. doi: 10.3934/publichealth.2019.4.568







 DownLoad:
DownLoad: