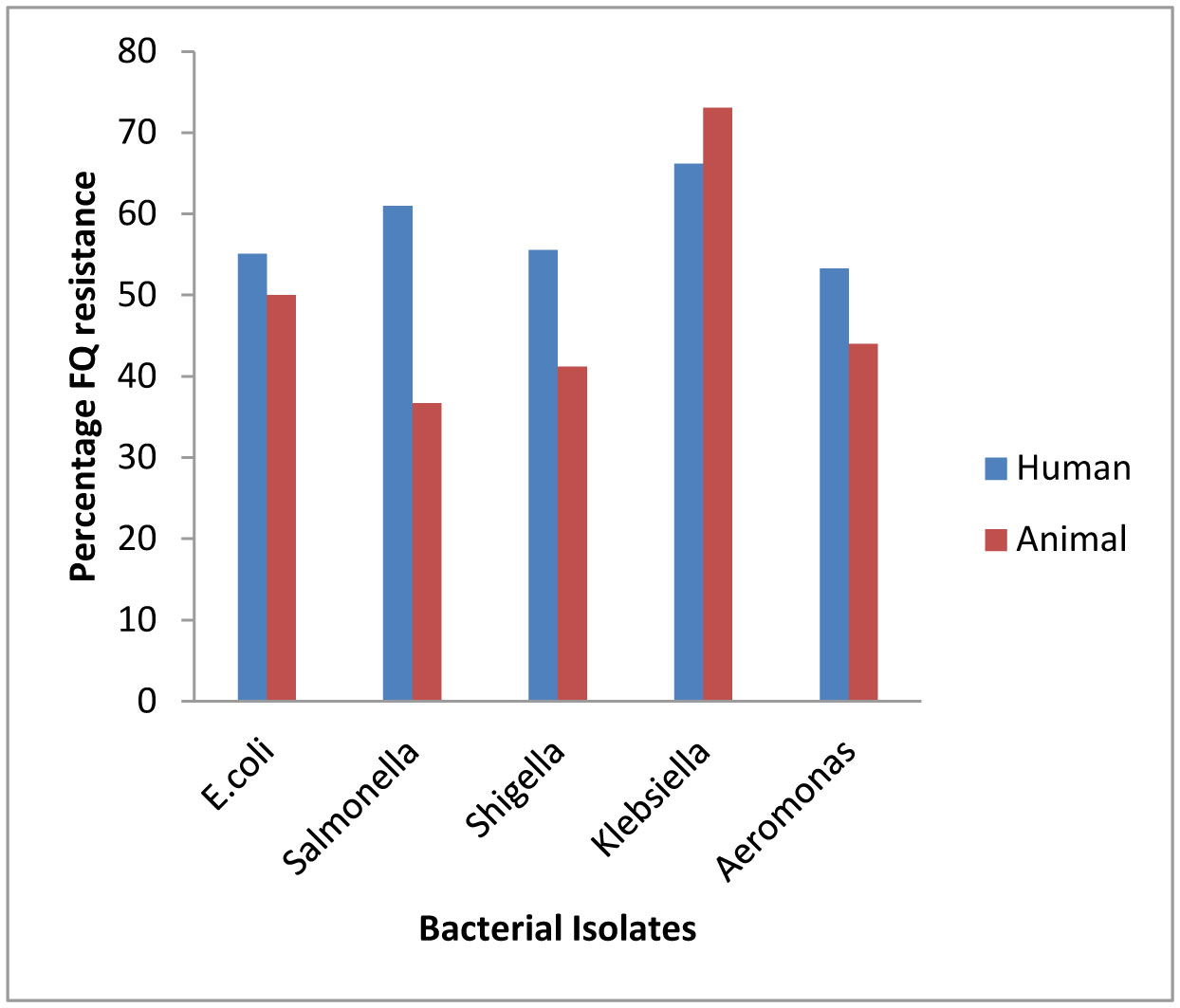
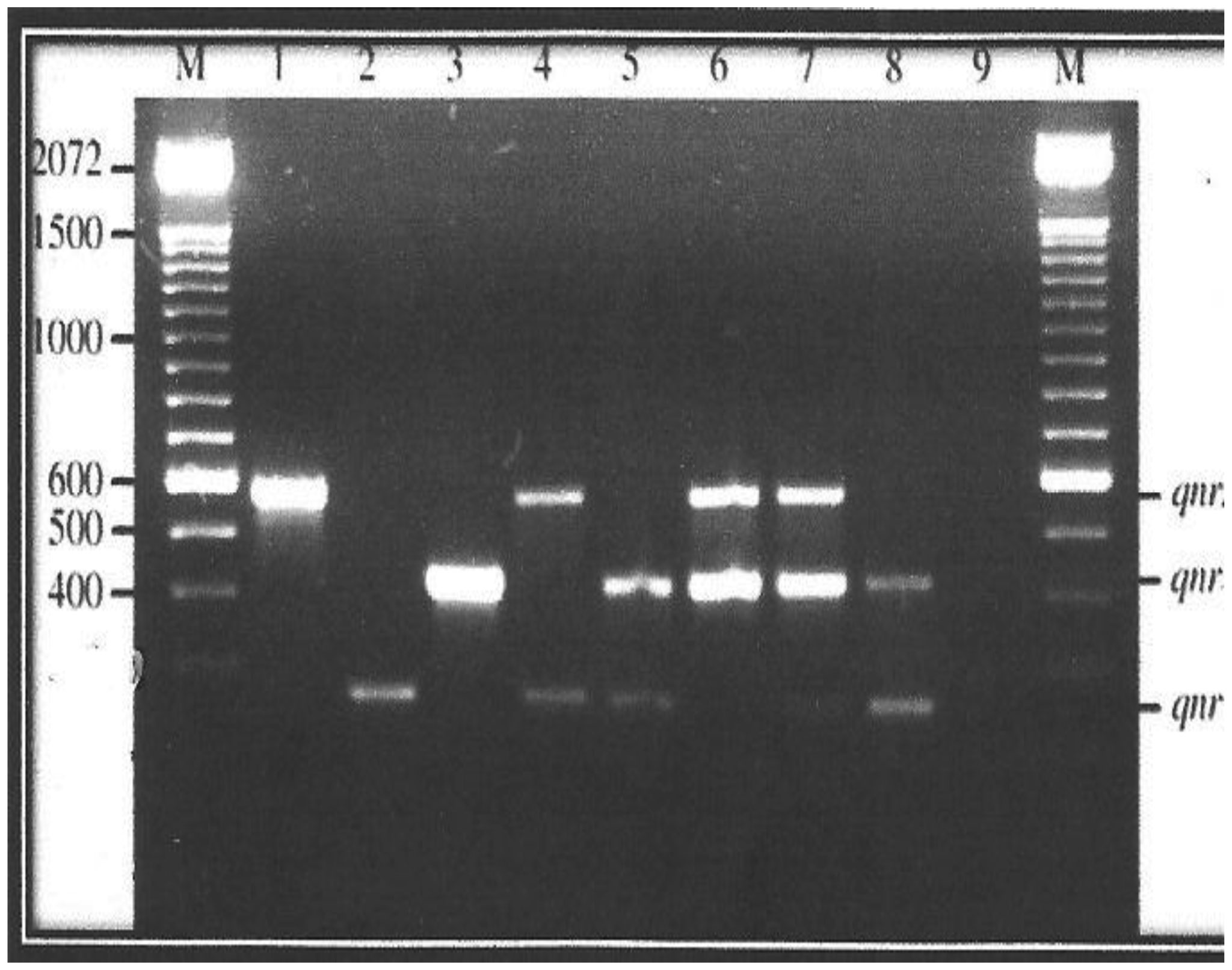
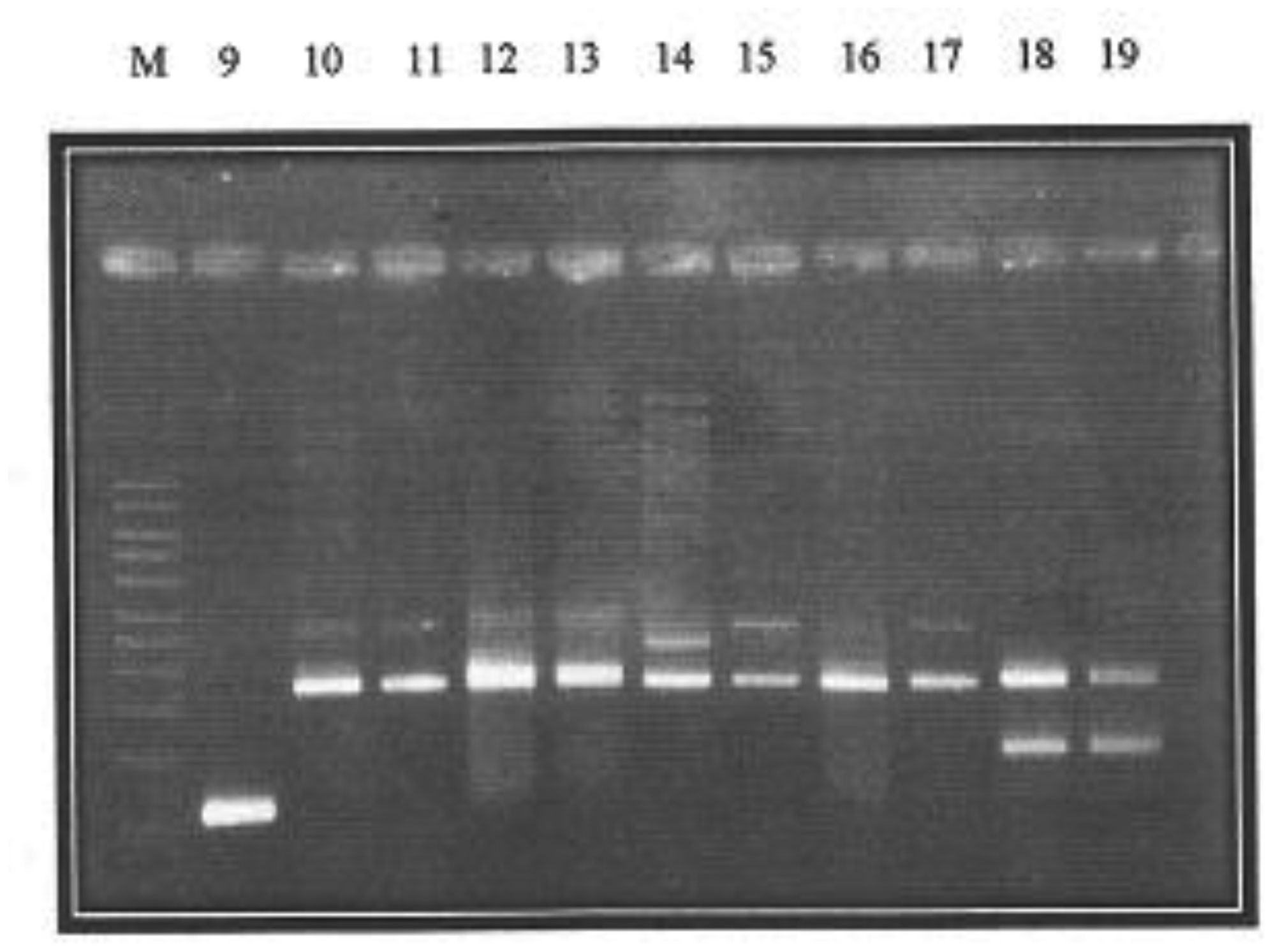
Plasmid mediated quinolone resistance (PMQR) is a public health challenge arising among other things, from indiscriminate use of the floroquinolones (FQr) prophylactically in animal husbandry. This study examines the occurrence of PMQR genes amongst enteric bacteria isolated from human and animal sources. A total of 720 (360 stool and 360 fish pond water/poultry litter) samples were examined for fluoroquinolone resistant (FQr) bacteria. Percentage FQr was generally higher among human isolates than isolates from animals. Proportion of PMQR amongst FQr isolates were (1.05 and 4.32) % for E. coli from human and animal sources. For Salmonella spp., Shigella spp., Klebsiella spp. and Aeromonas spp., percentages PMQR were 0.00 & 6.93, 0.00 & 6.38, 4.26 & 5.26 and 0.00 &3.03 for human and animal sources respectively, for the isolates. The PMQR genes: qnrA, qnr B, qnr S and qep A were 11, 15, 7 and 1 amongst a total of 1018 FQr and 29 PMQR isolates respectively. The aac (6′)–Ib-cr gene was not detected in this study. Approximate Plasmid bands of PCR amplicon for qnr A, qnr B, qnr S and qep A respectively were established. The proportion of PMQR genes especially among isolates from animal sources is of public health concern due to the higher possibility of a horizontal FQ resistance transfer to humans.
Citation: EHWARIEME Daniel Ayobola, WHILIKI Onoriadjeren Oscar, EJUKONEMU Francis Ejovwokoghene. Occurrence of plasmid mediated fluoroquinolone resistance genes amongst enteric bacteria isolated from human and animal sources in Delta State, Nigeria[J]. AIMS Microbiology, 2021, 7(1): 75-95. doi: 10.3934/microbiol.2021006
Plasmid mediated quinolone resistance (PMQR) is a public health challenge arising among other things, from indiscriminate use of the floroquinolones (FQr) prophylactically in animal husbandry. This study examines the occurrence of PMQR genes amongst enteric bacteria isolated from human and animal sources. A total of 720 (360 stool and 360 fish pond water/poultry litter) samples were examined for fluoroquinolone resistant (FQr) bacteria. Percentage FQr was generally higher among human isolates than isolates from animals. Proportion of PMQR amongst FQr isolates were (1.05 and 4.32) % for E. coli from human and animal sources. For Salmonella spp., Shigella spp., Klebsiella spp. and Aeromonas spp., percentages PMQR were 0.00 & 6.93, 0.00 & 6.38, 4.26 & 5.26 and 0.00 &3.03 for human and animal sources respectively, for the isolates. The PMQR genes: qnrA, qnr B, qnr S and qep A were 11, 15, 7 and 1 amongst a total of 1018 FQr and 29 PMQR isolates respectively. The aac (6′)–Ib-cr gene was not detected in this study. Approximate Plasmid bands of PCR amplicon for qnr A, qnr B, qnr S and qep A respectively were established. The proportion of PMQR genes especially among isolates from animal sources is of public health concern due to the higher possibility of a horizontal FQ resistance transfer to humans.

| [1] | Crump P, Lubsy JA, Mintz SP (2004) The global burden of enteric fever. Bull World Health Organ 82: 346-353. |
| [2] | Al-Sanouri TM, Paglietti B, Haddadin A, et al. (2008) Emergence of plasmid-mediated multidrug resistance in epidemic and non-epidemic strains of Salmonella enteric serotype Typhi from Jordan. J Infect Dev Countries 2: 295-301. |
| [3] | Octavia S, Lan R (2014) The family Enterobacteriaceae. The Prokaryotes Berlin, Heidelberg: Springer, 225-286. |
| [4] | Oliveira RV, Oliveira MC, Pelli A (2017) Disease infection by Enterobacteriaceae family in fishes: A review. J Microbiol Exp 4: 001128. |
| [5] | Okafor N, Okeke BC (2007) Modern industrial microbiology and biotechnology Enfield, New Hamshire, United States of America: Science Publishers, 530. |
| [6] |
Ball P (2000) Quinolone generations: natural history or natural selection? J Antimicrob Chemother 46: 17-24. doi: 10.1093/oxfordjournals.jac.a020889

|
| [7] |
Pharm TDM, Ziora ZM, Blaskovich MAT (2019) Quinolone antibiotics. Med Chem Comm 10: 1719-1739. doi: 10.1039/C9MD00120D
|
| [8] | Oliphant CM, Green GM (2002) Quinolones: A comprehensive review. Am Fam Physician 65: 455-464. |
| [9] |
Nuesch-Inderbinen M, Abgottspon H, Sagessa G, et al. (2015) Antimicrobial susceptibility of travel–related Salmonella enteric serovartyphi isolates detected in Switzerland (2002–2013) and molecular characterization of quinolone resistant isolates. BMC Infect Dis 15: 212. doi: 10.1186/s12879-015-0948-2
|
| [10] | Choffness E, Pelman D, Mark A (2011) Antibiotic resistance: implication for global health and novel intervention Washinghton DC: National Academies Press. |
| [11] |
Prestinaci F, Pezzotti P, Pantosti A (2015) Antimicrobial resistance: a global multifaceted phenomenon. Pathog Glob Health 109: 309. doi: 10.1179/2047773215Y.0000000030
|
| [12] |
Shrestha P, Cooper B.S, Coast J, et al. (2018) Enumerating the economic cost of antimicrobial resistance per antibiotic consumed to inform the evaluations of interventions affecting their use. Antimicrob Resit Infect Control 7: 98. doi: 10.1186/s13756-018-0384-3
|
| [13] | Dadgostar P (2019) Antimicrobial resistance: Implications and costs. Dove Medical Press 12: 3903-3910. |
| [14] |
Andersson DI, Hughes D (2014) Microbiological effects of sublethal levels of antibiotics. Nat Rev Microbiol 12: 465-478. doi: 10.1038/nrmicro3270
|
| [15] | Woolhouse M, Ward M, van Bunnik B, et al. (2015) Antimicrobial resistance in humans, livestock and the wider environment. Philos Trans R Soc B370: 201400683. |
| [16] |
Marshall BM, Levy SB (2011) Food animals and antimicrobials: Impacts on human health. Clin Microbiol Rev 24: 718-733. doi: 10.1128/CMR.00002-11
|
| [17] |
Yang H, Duan G, Zhu J, et al. (2013) Prevalence and characterization of plasmid–mediated quinolone resistance and mutations in the gyrase and topoisomerase IV genes among Shigella isolates from Henan, China between 2001 and 2008. Int J Antimicrob Agents 42: 173-177. doi: 10.1016/j.ijantimicag.2013.04.026
|
| [18] |
Jacoby GA, Strahilevitz J, Hooper DC (2014) Plasmid–mediate quinolone resistance. Microbiol Spectrum 2: 0006-2013. doi: 10.1128/microbiolspec.PLAS-0006-2013
|
| [19] |
Poirel L, Cattoir V, Nordmann P (2012) Plasmid–Mediated quinolone resitance interractions between human, animal and environmental ecologies. Front Microbiol 3: 24. doi: 10.3389/fmicb.2012.00024
|
| [20] | Murray PR, Baron JE, Jorgensen JH, et al. (2008) Manual of clinical microbiology. Clini Infect Diss 46. |
| [21] | Clinical and laboratory standard institute (CLSI) (2014) Performance Standards for Antimicrobial Disc Susceptibility Test, Twenty-third information supplement. CLSI Document 33: M100-S23. |
| [22] | Silhavey TJ, Berman ML, Enquist LW (1984) Experiments with gene fusions Cold Spring Harbor, NY: Cold Spring Harbor Laboratory press. |
| [23] |
Elnifro EM, Ashshi AM, Cooper RJ, et al. (2000) Multiplex PCR: optimization and application in diagnostic virology. Clin Microbiol Rev 13: 559-570. doi: 10.1128/CMR.13.4.559
|
| [24] |
Robicsek A, Strahilevitz J, Sham DF, et al. (2006) qnr prevalence in Ceftazidime-resistant Enterobacteriaceae isolated from the United States. Antimicrob Agents Chemother 50: 2872-2874. doi: 10.1128/AAC.01647-05
|
| [25] |
Park D, Hyun J, Park Y, et al. (2006) Sensitive and specific detection of Xanthomonas axonopodis pv. Citri by PCR using pathovar specific primers based on hrpW gene sequences. Microbiol Res 161: 145-149. doi: 10.1016/j.micres.2005.07.005
|
| [26] |
Yamane K, Wachino J, Suzuki S, et al. (2008) Plasmid-mediated qepA gene among Escherichia coli clinical isolates from Japan. Antimicrob Agents Chemother 52: 1564-1566. doi: 10.1128/AAC.01137-07
|
| [27] | Romero J, Feijoo CG, Navarrete P (2012) Antibiotics in aquaculture–use, abuse and alternatives. Health and environment in aquaculture Intech open. |
| [28] |
Barbosa MMC, Pinto FR, Ribeiro LF, et al. (2014) Serology and patterns of antimicrobial susceptibility in Escherichia coli isolates from pay-to-fish ponds. Arq Inst Biol São Paulo 81: 43-48. doi: 10.1590/S1808-16572014000100008
|
| [29] |
Chen Z, Jiang X (2014) Microbiological Safety of Chicken litter or chicken litter–based organic fertilizers: A review. Agriculture 4: 1-29. doi: 10.3390/agriculture4010001
|
| [30] |
Chinivasagam HN, Redding M, Runge G, et al. (2010) Presence and incidence of food-borne pathogens in Australian Chicken litter. Br Poult Sci 51: 311-318. doi: 10.1080/00071668.2010.499424
|
| [31] |
Padmavathy K, Krishnan P, Rajajekaran S (2014) Fluoroquinolone resistance among CTM-M producing Uropathogenic Escherichia coli from non-HIV patients in South India. BMC Infect Dis 14: 63. doi: 10.1186/1471-2334-14-S3-P63
|
| [32] |
Asensio A, Alvarez-Espejo T, Fernandez-Crehuet J, et al. (2011) Trends in yearly prevalence of third-generation cephalosporin and fluoroquinolone resistant Enterobacteriaceae infections and antimicrobial use in Spanish hospitals, Spain, 1999 to 2010. Eur Surveill 16: 19983. doi: 10.2807/ese.16.40.19983-en
|
| [33] |
Dalhoff A (2012) Global fluoroquinolone resistance epidemiology and implications for clinical use. Interdiscip Perspect Infect Dis 2012: 37. doi: 10.1155/2012/976273
|
| [34] |
Robicsek A, Strahilevitz J, Jacoby GA, et al. (2006) Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med 12: 83-88. doi: 10.1038/nm1347
|
| [35] | Lavanya B, Sowmiya S, Balaji S, et al. (2011) Plasmid profiling and curing of Lactobacillus strains isolated from fermented milk for probiotic applications. Adv J Food Technol 3: 95-101. |
| [36] |
Salisbury J, Hedges RM, Datta N (1972) Two methods of curing transmissible bacterial plasmids. J Gen Microbiol 70: 443-452. doi: 10.1099/00221287-70-3-443
|
| [37] |
Chen X, Zhang W, Pan W, et al. (2012) Prevelence of qnr,aac(6′)-Ib-cr, qepA and oqxAB in Escherichia coli isolated from humans, animals and the environment. Antimicrob Agents Chemother 56: 3423-3427. doi: 10.1128/AAC.06191-11
|
| [38] |
Pitout JD, Gregson DB, Campbell L, et al. (2009) Molecular characteristics of extended-spectrum-betalactamase-producing Escherichia coli isolates using bacteremia in the Calgary Health Region from 2000 to 2007: emergence of clone ST131 as a cause of community-acquired infections. Antimicrob Agents Chemother 53: 2846-2851. doi: 10.1128/AAC.00247-09
|
| [39] |
Jiang Y, Zhou Z, Qian Y, et al. (2008) Plasmid-mediated quinolone resistance determinants qnr and aac(6′)-Ib-cr in extended-spectrum b-lactamase-producing Escherichia coli and Klebsiella pneumoniae in China. J Antimicrob Chemother 61: 1003-1006. doi: 10.1093/jac/dkn063
|
| [40] |
Guan X, Xue X, Liu Y, et al. (2013) Plasmid-mediated quinolone resistance- current knowledge and fatal perspectives. J Int Med Res 41: 20-30. doi: 10.1177/0300060513475965
|
| [41] |
Yamane K, Wachino J, Suzuki S, et al. (2007) New plasmid mediated fluoroquinolone efflux pump, QepA, found in an Escherichia coli clinical isolate. Antimicrob Agents Chemother 51: 3354-3360. doi: 10.1128/AAC.00339-07
|
| [42] |
Vien TML, Minh NNQ, Thuong TC, et al. (2012) The Selection of Fluoroquinolone Resistant genes in the gut flora of Vietnamese children. PLoS One 7: e42919. doi: 10.1371/journal.pone.0042919
|
| [43] | Chen PL, Wu CJ, Chang CM, et al. (2007) Extra-intestinal focal infections in adults with Salmonella enterica serotype Choleraesuisbacteraemia. J Microbiol Immunol Infect 40: 240-247. |
| [44] |
Cattoir V, Poirel L, Nordmann P (2007) Plasmid-mediated quinolone resistance determinant qnrB4 in France from an Enterobacter cloacae clinical isolate coexpressing a qnrS1 determinant. Antimicrob Agents Chemother 51: 2652-2653. doi: 10.1128/AAC.01616-06
|
| [45] |
Liu JH, Deng YT, Zeng ZL, et al. (2008) Coprevalence of plasmid-mediated quinolone resistance determinanats qepA, qnr and aac(6′)-Ib-cr among 16S rRNA Methylase RmtB-producing Escherichia coli isolated from pigs. Antimicrobl Agents Chemother 52: 2992-2993. doi: 10.1128/AAC.01686-07
|
| [46] |
Ma J, Zeng Z, Chen Z, et al. (2009) High prevalence of plasmid-mediated quinolone resistance determinants qnr, aac(6′)-Ib-cr and qepA among ceftiofur-resistant Enterobacteriaceae isolates from companion and food-producing animals. Antimicrob Agents Chemother 53: 519-524. doi: 10.1128/AAC.00886-08
|
| [47] | Kim J, Luo F, Jiang X (2009) Factors impacting the growth of Esherichia coli 0157:H7 in dairy manure compost. J Food Protect 72: 157601584. |
| [48] | Chong YP, Jun J, Yoon HJ, et al. (2007) Prevalence of aac(6′)-Ib-cr encoding a Ciprofloxacin-modifying enzyme in Enterobacteriacea isolated on blood cultures in Korea. Abstracts of Forty-seventh Interscience Conference on Antimicrobial Agents and Chemotherapy American Society of Microbiology, C2-154. |
| [49] |
Park CH, Robicsek A, Jacoby GA, et al. (2006) Prevalence in the United States of aac(6′)-Ib-cr encoding a ciprofloxacin–modifying enzyme. Antimicrob Agents Chemother 50: 3953-3955. doi: 10.1128/AAC.00915-06
|
| [50] |
Sjolund-Karlsson M, Folster JP, Pecic G, et al. (2009) Emergence of Plasmid-mediated Quinolone Resistance among Non-Typhi Salmonella enteric isolates from Humans in the United States. Antimicrob Agents Chemother 53: 2142-2144. doi: 10.1128/AAC.01288-08
|
| [51] |
Cavaco LM, Hasman H, Xia S, et al. (2009) qnrD, a novel gene conferring transferable quinolone resistance in Salmonella enterica Serovar Kentucky and Bovismorbificans strains of human origin. Antimicrob Agents Chemother 53: 603-608. doi: 10.1128/AAC.00997-08
|
| [52] |
Poirel L, Leviandier C, Nordmann P (2006) Prevalence and genetic analysis of plasmid-mediated quinolone resistance determinants qnrA and qnrS in Enterobacteriacae isolates from a French university hospital. Antimicrob Agents Chemother 50: 3992-3997. doi: 10.1128/AAC.00597-06
|
| [53] |
Lavilla S, Gonzalez-lopez JJ, Sabate M, et al. (2008) Prevelence of qnr genes among extended-spectrum β- lactamase producing enterobacterial isolates in Barcelona. J Antimicrob Chemother 61: 291-295. doi: 10.1093/jac/dkm448
|
| [54] |
Tran JH, Jacoby GA (2002) Mechanism of plasmid-mediated quinolone resistance. Proceedings of the National Academy of Science 99: 5638-5642. doi: 10.1073/pnas.082092899
|
| [55] | Cano ME, Martinez-Martinez L, Garcia-Lobo JM, et al. (2005) Detection of orf513 and qnrA among multiresistant gram-negative clinical isolates in Spain, abstr. C1-1043/69. Abstr. 45th Intersci. Conf. Antimicrob. Agents Chemother Washington, DC: American Society for Microbiology. |
| [56] |
Hopkins KL, Davies RH, Threlfall EJ (2005) Mechanisms of quinolone resistance in Escherichia coli and Salmonella: Recent developments. Int J Antimicrob Agents 25: 358-373. doi: 10.1016/j.ijantimicag.2005.02.006
|
| [57] |
Corkill JE, Anson JJ, Hart CA (2005) High prevalence Plasmid-mediated quinolone resistance determinants qnrA in multidrug-resistant Enterobacteria from blood culture. J Antimicroial Chemother 56: 1115-1117. doi: 10.1093/jac/dki388
|
Figures(3) / Tables(11)

EHWARIEME Daniel Ayobola, WHILIKI Onoriadjeren Oscar, EJUKONEMU Francis Ejovwokoghene. Occurrence of plasmid mediated fluoroquinolone resistance genes amongst enteric bacteria isolated from human and animal sources in Delta State, Nigeria[J]. AIMS Microbiology, 2021, 7(1): 75-95. doi: 10.3934/microbiol.2021006







 DownLoad:
DownLoad: