Citation: Hend Amraoui, Faouzi Mhamdi, Mourad Elloumi. Survey of Metaheuristics and Statistical Methods for Multifactorial Diseases Analyses[J]. AIMS Medical Science, 2017, 4(3): 291-331. doi: 10.3934/medsci.2017.3.291

| [1] |
Campion D (2001) Dissection génétique des maladies à hérédité complexe'. médecine/sciences 17: 1139-1148. doi: 10.1051/medsci/200117111139

|
| [2] | Hodge SE, Hager VR, Greenberg DA (2016) Using Linkage Analysis to Detect Gene-Gene Interactions. 2. Improved Reliability and Extension to More-Complex Models', Plos one 11: e0146240. |
| [3] | Onouchi Y (2017) Identification of Novel Kawasaki Disease Susceptibility Genes by Genome-Wide Association Studies. In: Kawasaki Disease. Springer Japan: 23-29. |
| [4] | Morton NE (1955) Sequential tests for the detection of linkage. Am J Hum Genet 7: 277. |
| [5] | Clerget-Darpoux F, Bonaiti-Pellie C, Hochez J (1986) Effects of misspecifying genetic parameters in lod score analysis. Biometrics: 393-399. |
| [6] | Savard N (2005) Méthode d'analyse de liaison génétique pour des familles dans lesquelles il ya de l'hétérogénéité non-allélique intra-familiale. Université Laval. |
| [7] |
Haseman J, Elston R (1972) The investigation of linkage between a quantitative trait and a marker locus. Behav Genet 2: 3-19. doi: 10.1007/BF01066731
|
| [8] |
Costantino F, Chaplais E, Leturcq T, et al. (2016) Whole-genome single nucleotide polymorphism-based linkage analysis in spondyloarthritis multiplex families reveals a new susceptibility locus in 13q13. Ann Rheum Dis 75: 1380-1385. doi: 10.1136/annrheumdis-2015-207720
|
| [9] |
Hu H., Roach JC, Coon H, et al. (2014) A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat Biotechnol 32: 663-669. doi: 10.1038/nbt.2895
|
| [10] |
Greenwood TA, Swerdlow NR, Gur RE, et al. (2013) Genome-wide linkage analyses of 12 endophenotypes for schizophrenia from the Consortium on the Genetics of Schizophrenia. Am J Psychiat 170: 521-532. doi: 10.1176/appi.ajp.2012.12020186
|
| [11] |
Cochran WG (1954) Some methods for strengthening the common χ 2 tests'. Biometrics 10: 417-451. doi: 10.2307/3001616
|
| [12] |
Armitage P (1955) Tests for linear trends in proportions and frequencies. Biometrics 11: 375-386. doi: 10.2307/3001775
|
| [13] | Wang L, Ni H, Yang R, et al. (2013) Feature selection based on meta-heuristics for biomedicine. Optim Method Softw 29: 703-719. |
| [14] | Talbi EG, Jourdan L, Garcia-Nieto J, et al. (2008) Comparison of population based metaheuristics for feature selection: Application to microarray data classification. Computer Systems and Applications. AICCSA 2008. IEEE/ACS International Conference on. IEEE: 45-52. |
| [15] |
Yusta SC (2009) Different metaheuristic strategies to solve the feature selection problem. Pattern Recogn Lett 30: 525-534. doi: 10.1016/j.patrec.2008.11.012
|
| [16] | Amarnath B, alias Balamurugan SA (2016) Metaheuristic Approach for Efficient Feature Selection: A Data Classification Perspective. Indian J Sci Technol 9. |
| [17] | Carrizosa E, Martin-Barragan B, Romero Morales D (2012) Variable neighborhood search for parameter tuning in support vector machines. Tech. rep. |
| [18] | Chan K, Zhu H, Aydin M, et al. (2008) An integrated approach of support vector machine and variable neighborhood search for discovering combinational gene signatures in predicting chemo-response of osteosarcoma. Proceedings of the international multiconference of engineers and computer scientists. 1: 121-125. |
| [19] |
García-Torres M, Gómez-Vela F, Melián-Batista B, et al. (2016) High-dimensional feature selection via feature grouping: A Variable Neighborhood Search approach. Inform Sciences 326: 102-118. doi: 10.1016/j.ins.2015.07.041
|
| [20] |
Belacel N, Čuperlović-Culf M, Laflamme M, et al. (2004) Fuzzy J-Means and VNS methods for clustering genes from microarray data. Bioinformatics 20: 1690-1701. doi: 10.1093/bioinformatics/bth142
|
| [21] |
Cahon S, Melab N, Talbi EG (2004) Paradiseo: A framework for the reusable design of parallel and distributed metaheuristics. J Heuristics 10: 357-380. doi: 10.1023/B:HEUR.0000026900.92269.ec
|
| [22] |
García-Torres M, Armañanzas R, Bielza C, et al. (2013) Comparison of metaheuristic strategies for peakbin selection in proteomic mass spectrometry data. Inform Sciences 222: 229-246. doi: 10.1016/j.ins.2010.12.013
|
| [23] |
Feo TA, Resende MGC, Smith SH (1994) A Greedy Randomized Adaptive Search Procedure for Maximum Independent Set. Oper Res 42: 860-878. doi: 10.1287/opre.42.5.860
|
| [24] |
Kirkpatrick S, Gelatt CD, Vecchi MP (1983) Optimization by Simulated Annealing. Science 220: 671-680. doi: 10.1126/science.220.4598.671
|
| [25] |
Glover F (1989) Tabu search-part I. ORSA Journal on computing 1: 190-206. doi: 10.1287/ijoc.1.3.190
|
| [26] | Papadimitriou CH, Steiglitz K (1982) Combinatorial optimization: algorithms and complexity. Courier Corporation. |
| [27] | Papadimitriou CH (1976) The complexity of combinatorial optimization problems. |
| [28] | Schlosberg CE, Schwantes-An TH, Duan W, et al. (2011) Application of Bayesian network structure learning to identify causal variant SNPs from resequencing data. BMC Proceedings 5 Suppl 9: S109-S109. |
| [29] | Nithya R, Venkateswaran N (2015) Analysis of Segmentation Algorithms in Colour Fundus and OCT Images for Glaucoma Detection. Indian J Sci Technol 8. |
| [30] | Bhardwaj A, Tiwari A, Varma MV, et al. (2015) An Analysis of Integration of Hill Climbing in Crossover and Mutation operation for EEG Signal Classification. Proceedings of the 2015 Annual Conference on Genetic and Evolutionary Computation. ACM: 209-216. |
| [31] |
Metropolis N, Rosenbluth AW, Rosenbluth MN, et al. (1953) Equation of state calculations by fast computing machines. J Chem Phys 21: 1087-1092. doi: 10.1063/1.1699114
|
| [32] |
Iossifov I, Zheng T, Baron M, et al. (2008) Genetic-linkage mapping of complex hereditary disorders to a whole-genome molecular-interaction network. Genome Res 18: 1150-1162. doi: 10.1101/gr.075622.107
|
| [33] |
Wirdefeldt K, Burgess CE, Westerberg L, et al. (2003) A linkage study of candidate loci in familial Parkinson's Disease. BMC Neurol 3: 6. doi: 10.1186/1471-2377-3-6
|
| [34] |
Sartakhti JS, Zangooei MH, Mozafari K (2012) Hepatitis disease diagnosis using a novel hybrid method based on support vector machine and simulated annealing (SVM-SA). Comput Meth Prog Bio 108: 570-579. doi: 10.1016/j.cmpb.2011.08.003
|
| [35] | Goyal M, Dhanjal JK, Goyal S, et al. (2014) Development of dual inhibitors against Alzheimer's disease using fragment-based QSAR and molecular docking. Bio med Res Int: 2014. |
| [36] |
Glover F (1986) Future paths for integer programming and links to artificial intelligence. Comput Oper Res 13: 533-549. doi: 10.1016/0305-0548(86)90048-1
|
| [37] |
Glover F (1977) Heuristics for integer programming using surrogate constraints. Decision Sci 8: 156-166. doi: 10.1111/j.1540-5915.1977.tb01074.x
|
| [38] |
Shen Q, Shi WM, Kong W (2008) Hybrid particle swarm optimization and tabu search approach for selecting genes for tumor classification using gene expression data. Comput Biol Chem 32: 53-60. doi: 10.1016/j.compbiolchem.2007.10.001
|
| [39] | Wang S, Kong W, Zeng W, et al (2016) Hybrid Binary Imperialist Competition Algorithm and Tabu Search Approach for Feature Selection Using Gene Expression Data. Biomed Res Int: 2016. |
| [40] |
Nguyen T, Khosravi A, Creighton D, et al. (2015) Fuzzy system with tabu search learning for classification of motor imagery data. Biomed Signal Proces 20: 61-70. doi: 10.1016/j.bspc.2015.04.007
|
| [41] | Darwin C (1968) On the origin of species by means of natural selection. 1859. London: Murray Google Scholar. |
| [42] | Holland JH (1992) Adaptation in natural and artificial systems: an introductory analysis with applications to biology, control, and artificial intelligence. MIT press:1992. |
| [43] | Goldberg DE (1989) Genetic Algorithms in Search, Optimization, and Machine Learning. |
| [44] | Goldberg DE, Deb K (1991) A comparative analysis of selection schemes used in genetic algorithms. Foundations of Genetic Algorithms 1: 69-93. |
| [45] | Blickle T, Thiele L (1995) A Mathematical Analysis of Tournament Selection. ICGA: 9-16. |
| [46] | Sergii K, Yurii S, Tatyana V, et al. (2016) Feature Selection for Time-Series Prediction in Case of Undetermined Estimation. In: Biologically Inspired Cognitive Architectures (BICA) for Young Scientists. Springer, Cham: 85-97. |
| [47] |
Underwood DJ, Zhang J, Denton BT, et al. (2012) Simulation optimization of PSA-threshold based prostate cancer screening policies. Health Care Manag Sci 15: 293-309. doi: 10.1007/s10729-012-9195-x
|
| [48] |
Beheshti I, Demirel H, Matsuda H, et al. (2017) Classification of Alzheimer's disease and prediction of mild cognitive impairment-to-Alzheimer's conversion from structural magnetic resource imaging using feature ranking and a genetic algorithm. Comput Biol Med 83: 109-119. doi: 10.1016/j.compbiomed.2017.02.011
|
| [49] | Singh J, Kaur R (2016) Cardio Vascular Disease Classification Ensemble Optimization using Genetic Algorithm and Neural Network. Indian J Sci Technol 9: S1. |
| [50] | Paul AK, Shill PC, Rabin MRI, et al. (2016) Genetic algorithm based fuzzy decision support system for the diagnosis of heart disease. In: Informatics, Electronics and Vision (ICIEV), 2016 5th International Conference on. IEEE: 145-150. |
| [51] | Sachnev V, Suresh S, Choi YS (2016) Bio-marker Detector and Parkinson's disease diagnosis Approach based on Samples Balanced Genetic Algorithm and Extreme Learning Machine. 한국디지털콘텐츠학회논문지 17: 509-521. |
| [52] | Paul D, Su R, Romain M, et al. (2016) Feature selection for outcome prediction in oesophageal cancer using genetic algorithm and random forest classifier. Comput Med Imag Grap. |
| [53] |
López FG, Torres MG, Batista BM, et al. (2006) Solving feature subset selection problem by a parallel scatter search. Eur J Oper Res 169: 477-489. doi: 10.1016/j.ejor.2004.08.010
|
| [54] |
Nepomuceno JA, Troncoso A, Aguilar-Ruiz JS (2015) Scatter search-based identification of local patterns with positive and negative correlations in gene expression data. Appl Soft Comput 35: 637-651. doi: 10.1016/j.asoc.2015.06.019
|
| [55] | Lin SW, Chen SC (2012) Parameter determination and feature selection for C4. 5 algorithm using scatter search approach. Soft Comput 16: 63-75. |
| [56] |
Chen SC, Lin SW, Chou SY (2011) Enhancing the classification accuracy by scatter-search-based ensemble approach. Appl Soft Comput 11: 1021-1028. doi: 10.1016/j.asoc.2010.01.024
|
| [57] | Koza JR (1992) Genetic programming: on the programming of computers by means of natural selection, MIT press. |
| [58] | Sohn A, Olson RS, Moore JH (2017) Toward the automated analysis of complex diseases in genome-wide association studies using genetic programming. arXiv preprint arXiv: 1702.01780. |
| [59] | Vyas R, Bapat S, Goel P, et al. (2016) Application of Genetic Programming (GP) formalism for building disease predictive models from protein-protein interactions (PPI) data. IEEE/ACM Transactions on Computational Biology and Bioinformatics. |
| [60] | Hasan MK, Islam MM, Hashem M (2016) Mathematical model development to detect breast cancer using multigene genetic programming. Informatics, Electronics and Vision (ICIEV), 2016 5th International Conference on. IEEE: 574-579. |
| [61] |
Deneubourg JL, Pasteels JM, Verhaeghe JC (1983) Probabilistic behaviour in ants: a strategy of errors?. J Theor Biol 105: 259-271. doi: 10.1016/S0022-5193(83)80007-1
|
| [62] |
Deneubourg JL, Goss S (1989) Collective patterns and decision-making. Ethol Ecol Evol 1: 295-311. doi: 10.1080/08927014.1989.9525500
|
| [63] |
Husain NP, Arisa NN, Rahayu PN, et al. (2017) Least Squares Support Vector Machines Parameter Optimization Based on Improved Ant Colony Algorithm For Hepatitis Diagnosis. Jurnal Ilmu Komputer dan Informasi 10: 43-49. doi: 10.21609/jiki.v10i1.428
|
| [64] | Asad AH, Azar AT, Hassanien AE (2017) A new heuristic function of ant colony system for retinal vessel segmentation. In: Medical Imaging: Concepts, Methodologies, Tools, and Applications. IGI Global: 2063-2081. |
| [65] | Eberhart R, Kennedy J (1995) A new optimizer using particle swarm theory. Micro Machine and Human Science, 1995. MHS'95., Proceedings of the Sixth International Symposium on. IEEE: 39-43. |
| [66] |
Sali R, Shavandi H, Sadeghi M (2016) A clinical decision support system based on support vector machine and binary particle swarm optimisation for cardiovascular disease diagnosis. Int J Data Min Bioin 15: 312-327. doi: 10.1504/IJDMB.2016.078150
|
| [67] | Shahsavari MK, Rashidi H, Bakhsh HR (2016) Efficient classification of Parkinson's disease using extreme learning machine and hybrid particle swarm optimization. Control, Instrumentation, and Automation (ICCIA), 2016 4th International Conference on. IEEE: 148-154. |
| [68] | Jothi N (2016) Prediction of Generalized Anxiety Disorder Using Particle Swarm Optimization. Advances in Information and Communication Technology: Proceedings of the International Conference, ICTA 2016. Springer, 538: 480. |
| [69] | Kumar GK (2016) An Optimized Particle Swarm Optimization based ANN Model for Clinical Disease Prediction. Indian J Sci Technol 9. |
| [70] |
Yang CH, Weng ZJ, Chuang LY, et al. (2017) Identification of SNP-SNP interaction for chronic dialysis patients. Comput Biol Med 83: 94-101. doi: 10.1016/j.compbiomed.2017.02.004
|
| [71] |
Gunasundari S, Janakiraman S, Meenambal S (2016) Velocity Bounded Boolean Particle Swarm Optimization for improved feature selection in liver and kidney disease diagnosis. Expert Syst Appl 56: 28-47. doi: 10.1016/j.eswa.2016.02.042
|
| [72] | Muthanantha Murugavel A, Ramakrishnan S (2014) Optimal feature selection using PSO with SVM for epileptic EEG classification. Int J Data Min Bioin 16: 343-358. |
| [73] |
Farmer JD, Packard NH, Perelson AS (1986) The immune system, adaptation, and machine learning. Physica D: Nonlinear Phenomena 22: 187-204. doi: 10.1016/0167-2789(86)90240-X
|
| [74] | De Castro LN, Timmis J (2002) Artificial immune systems: a new computational intelligence approach. Springer Science & Business Media. |
| [75] |
Polat K, Şahan S, Güneş S (2007) Automatic detection of heart disease using an artificial immune recognition system (AIRS) with fuzzy resource allocation mechanism and k-nn (nearest neighbour) based weighting preprocessing. Expert Syst Appl 32: 625-631. doi: 10.1016/j.eswa.2006.01.027
|
| [76] |
Chikh MA, Saidi M, Settouti N (2012) Diagnosis of diabetes diseases using an artificial immune recognition system2 (AIRS2) with fuzzy k-nearest neighbor. J Med Syst 36: 2721-2729. doi: 10.1007/s10916-011-9748-4
|
| [77] |
Zhao W, Davis CE (2011) A modified artificial immune system based pattern recognition approach-An application to clinical diagnostics. Artif Intell Med 52: 1-9. doi: 10.1016/j.artmed.2011.03.001
|
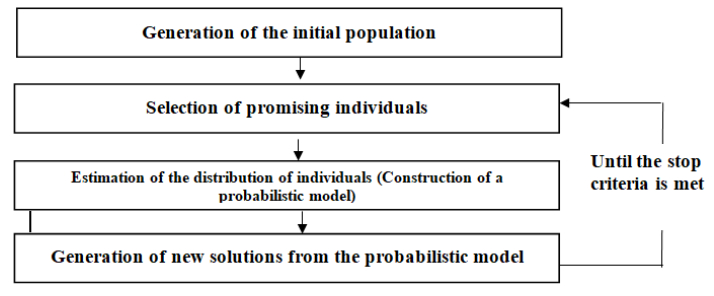
| [78] | Baluja S (1994) Population-based incremental learning. a method for integrating genetic search based function optimization and competitive learning. Carnegie-Mellon Univ Pittsburgh Pa Dept Of Computer Science. |
| [79] | Mühlenbein H, Paass G (1996) From recombination of genes to the estimation of distributions I. Binary parameters. Parallel problem solving from nature-PPSN IV: 178-187. |
| [80] |
Inza I, Merino M, Larrañaga P, et al. (2001) Feature subset selection by genetic algorithms and estimation of distribution algorithms: A case study in the survival of cirrhotic patients treated with TIPS. Artif Intell Med 23: 187-205. doi: 10.1016/S0933-3657(01)00085-9
|
| [81] |
Armananzas R, Saeys Y, Inza I, et al. (2011) Peakbin selection in mass spectrometry data using a consensus approach with estimation of distribution algorithms. IEEE/ACM Transactions on Computational Biology and Bioinformatics 8: 760-774. doi: 10.1109/TCBB.2010.18
|
| [82] |
Funayama M, Ohe K, Amo T, et al. (2015) CHCHD2 mutations in autosomal dominant late-onset Parkinson's disease: a genome-wide linkage and sequencing study. Lancet Neurol 14: 274-282. doi: 10.1016/S1474-4422(14)70266-2
|
| [83] |
Wotton CJ, Goldacre MJ (2014) Record-linkage studies of the coexistence of epilepsy and bipolar disorder. Soc Psych Psych Epid 49: 1483-1488. doi: 10.1007/s00127-014-0853-9
|
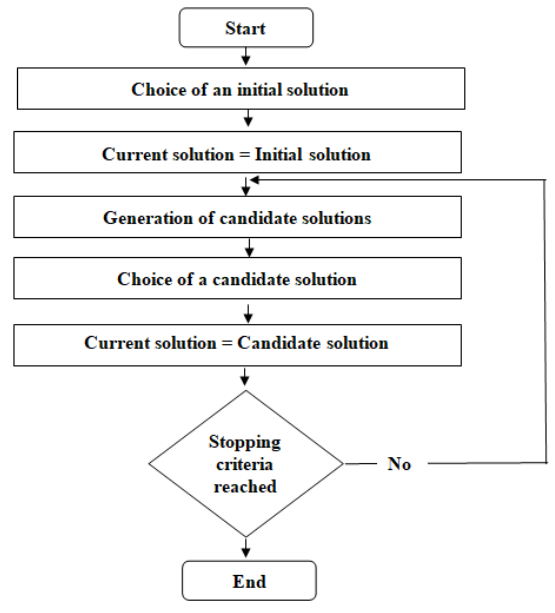
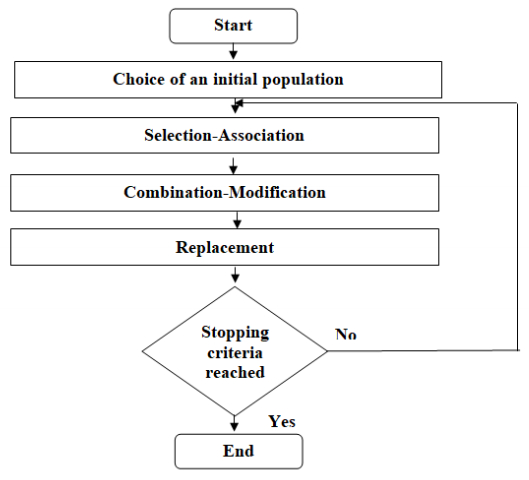
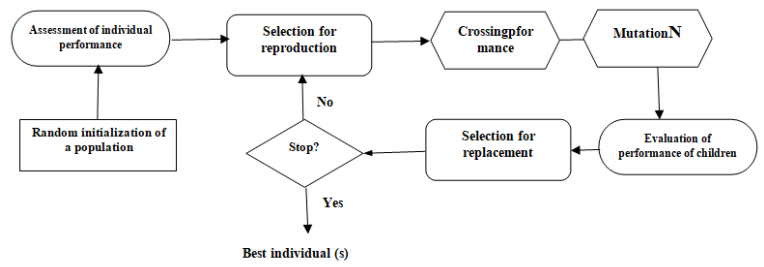
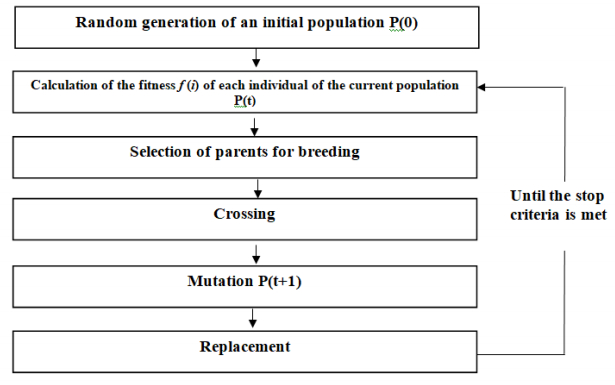
Figures(5) / Tables(5)

Hend Amraoui, Faouzi Mhamdi, Mourad Elloumi. Survey of Metaheuristics and Statistical Methods for Multifactorial Diseases Analyses[J]. AIMS Medical Science, 2017, 4(3): 291-331. doi: 10.3934/medsci.2017.3.291







 DownLoad:
DownLoad: