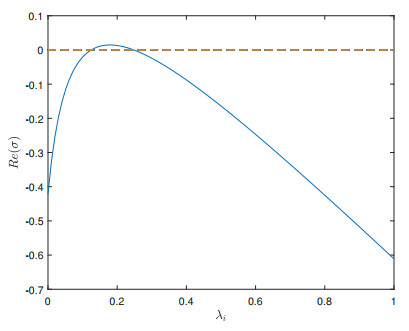
A vegetation model composed of water and plants was proposed by introducing a weighted graph Laplacian operator into the reaction-diffusion dynamics. We showed the global existence and uniqueness of the solution via monotone iterative sequence. The parameter space of Turing patterns for plant behavior is obtained based on the analysis of the eigenvalues of the Laplacian of weighted graph, while the amplitude equation determining the stability of Turing patterns is obtained by weakly nonlinear analysis. We also show that the optimal rainfall is only determined by the density of the water. By some numerical simulations, we examine the individual effect of diffusion term on the formation of regular Turing patterns. We show that the large diffusion induces stable Turing patterns.
Citation: Xiaomei Bao, Canrong Tian. Turing patterns in a networked vegetation model[J]. Mathematical Biosciences and Engineering, 2024, 21(11): 7601-7620. doi: 10.3934/mbe.2024334
A vegetation model composed of water and plants was proposed by introducing a weighted graph Laplacian operator into the reaction-diffusion dynamics. We showed the global existence and uniqueness of the solution via monotone iterative sequence. The parameter space of Turing patterns for plant behavior is obtained based on the analysis of the eigenvalues of the Laplacian of weighted graph, while the amplitude equation determining the stability of Turing patterns is obtained by weakly nonlinear analysis. We also show that the optimal rainfall is only determined by the density of the water. By some numerical simulations, we examine the individual effect of diffusion term on the formation of regular Turing patterns. We show that the large diffusion induces stable Turing patterns.

| [1] |
G. A. Worrall, Tree patterns in the Sudan, Eur. J. Soil Sci., 11 (1960), 63–67. https://doi.org/10.1111/j.1365-2389.1960.tb02202.x doi: 10.1111/j.1365-2389.1960.tb02202.x

|
| [2] |
L. P. White, Brousse tigree patterns in southern Niger, J. Ecol., 58 (1970), 549–553. https://doi.org/10.2307/2258290 doi: 10.2307/2258290
|
| [3] |
J. A. Ludwig, D. J. Tongway, Spatial organisation of landscapes and its function in semi-arid woodlands, Australia, Landscape Ecol., 10 (1995), 51–63. https://doi.org/10.1007/BF00158553 doi: 10.1007/BF00158553
|
| [4] |
C. Montana, J. Lopez-Portillo, A. Mauchamp, The response of two woody species to the conditions created by a shifting ecotone in an arid ecosystem, J. Ecol., 78 (1990), 789–798. https://doi.org/10.2307/2260899 doi: 10.2307/2260899
|
| [5] |
C. A. Klausmeier, Regular and irregular patterns in semiarid vegetation, Science, 284 (1999), 1826–1828. https://doi.org/10.1126/science.284.5421.1826 doi: 10.1126/science.284.5421.1826
|
| [6] |
R. Lefever, O. Lejeune, On the origin of tiger bush, Bull. Math. Biol., 59 (1997), 263–294. https://doi.org/10.1007/BF02462004 doi: 10.1007/BF02462004
|
| [7] |
J. A. Sherratt, An analysis of vegetative stripe formation in semi-arid landscape, J. Math. Biol., 51 (2005), 183–197. https://doi.org/10.1007/s00285-005-0319-5 doi: 10.1007/s00285-005-0319-5
|
| [8] |
B. J. Kealy, D. J. Wollkind, A nonlinear stability analysis of vegetative Turing pattern formation for an interaction-diffusion plant-surface water model system in an arid flat environment, Bull. Math. Biol., 74 (2012), 803–833. https://doi.org/10.1007/s11538-011-9688-7 doi: 10.1007/s11538-011-9688-7
|
| [9] |
Q. Xue, G. Sun, C. Liu, Z. Guo, Z. Jin, Y. Wu, et al., Spatiotemporal dynamics of a vegetation model with nonlocal delay in semi-arid environment, Nonlinear Dyn., 99 (2020), 3407–3420. https://doi.org/10.1007/s11071-020-05486-w doi: 10.1007/s11071-020-05486-w
|
| [10] |
Y. R. Zelnik, P. Gandhi, E. Knobloch, E. Meron, Implications of tristability in pattern-forming ecosystems, Chaos, 28 (2018), 033609. https://doi.org/10.1063/1.5018925 doi: 10.1063/1.5018925
|
| [11] |
P. Carter, A. Doelman, Traveling stripes in the Klausmeier model of vegetation pattern formation, SIAM J. Appl. Math., 78 (2018), 3213–3237. https://doi.org/10.1137/18M1196996 doi: 10.1137/18M1196996
|
| [12] |
R. Bastiaansen, P. Carter, A. Doelman, Stable planar vegetation stripe patterns on sloped terrain in dryland ecosystems, Nonlinearity, 32 (2019), 2759. https://doi.org/10.1088/1361-6544/ab1767 doi: 10.1088/1361-6544/ab1767
|
| [13] |
L. Eigentler, J. A. Sherratt, Long-range seed dispersal enables almost stationary patterns in a model for dryland vegetation, J. Math. Biol., 86 (2023), 15. https://doi.org/10.1007/s00285-022-01852-x doi: 10.1007/s00285-022-01852-x
|
| [14] |
R. Martinez-Garcia, C. Cabal, J. M. Calabrese, E. Hernández-García, C. E. Tarnita, C. López, et al., Integrating theory and experiments to link local mechanisms and ecosystem-level consequences of vegetation patterns in drylands, Chaos, Solitons Fractals, 166 (2023), 112881. https://doi.org/10.1016/j.chaos.2022.112881 doi: 10.1016/j.chaos.2022.112881
|
| [15] |
G. Consolo, G. Grifò, G. Valenti, Modeling vegetation patterning on sloped terrains: The role of toxic compounds, Physica D, 459 (2024), 134020. https://doi.org/10.1016/j.physd.2023.134020 doi: 10.1016/j.physd.2023.134020
|
| [16] |
G. Consolo, C. Currò, G. Grifò, G. Valenti, Stationary and oscillatory patterned solutions in three-compartment reaction–diffusion systems: Theory and application to dryland ecology, Chaos, Solitons Fractals, 186 (2024), 115287. https://doi.org/10.1016/j.chaos.2024.115287 doi: 10.1016/j.chaos.2024.115287
|
| [17] |
C. Tian, Turing pattern formation in a semiarid vegetation model with fractional-in-space diffusion, Bull. Math. Biol., 77 (2015), 2072–2085. https://doi.org/10.1007/s11538-015-0116-2 doi: 10.1007/s11538-015-0116-2
|
| [18] |
C. Tian, Z. Ling, L. Zhang, Delay-driven spatial patterns in a network-organized semiarid vegetation model, Appl. Math. Comput., 367 (2020), 124778. https://doi.org/10.1016/j.amc.2019.124778 doi: 10.1016/j.amc.2019.124778
|
| [19] |
G. Consolo, C. Currò, G. Valenti, Pattern formation and modulation in a hyperbolic vegetation model for semiarid environments, Appl. Math. Modell., 43 (2017), 372–392. https://doi.org/10.1016/j.apm.2016.11.031 doi: 10.1016/j.apm.2016.11.031
|
| [20] |
G. Grifò, G. Consolo, C. Currò, G. Valenti, Rhombic and hexagonal pattern formation in 2D hyperbolic reaction-transport systems in the context of dryland ecology, Physica D, 449 (2023), 133745. https://doi.org/10.1016/j.physd.2023.133745 doi: 10.1016/j.physd.2023.133745
|
| [21] |
G. Grifò, Vegetation patterns in the hyperbolic Klausmeier model with secondary seed dispersal, Mathematics, 11 (2023), 1084. https://doi.org/10.3390/math11051084 doi: 10.3390/math11051084
|
| [22] |
C. Currò, G. Grifò, G. Valenti, Turing patterns in hyperbolic reaction-transport vegetation models with cross-diffusion, Chaos, Solitons Fractals, 176 (2023), 114152. https://doi.org/10.1016/j.chaos.2023.114152 doi: 10.1016/j.chaos.2023.114152
|
| [23] |
H. Nakoa, A. S. Mikhailov, Turing patterns in network-oragnized activator-inhibitor systems, Nat. Phys., 6 (2010), 544–550. https://doi.org/10.1038/nphys1651 doi: 10.1038/nphys1651
|
| [24] |
J. Petit, T. Carletti, M. Asllani, D. Fanelli, Delay-induced Turing-like waves for one-species reaction-diffusion model on a network, Europhys. Lett., 111 (2015), 58002. https://doi.org/10.1209/0295-5075/111/58002 doi: 10.1209/0295-5075/111/58002
|
| [25] |
M. Banerjee, S. Petrovskii, Self-organised spatial patterns and chaos in a ratio-dependent predator-prey system, Theor. Ecol., 4 (2011), 37–53. https://doi.org/10.1007/s12080-010-0073-1 doi: 10.1007/s12080-010-0073-1
|
| [26] |
L. A. Segel, J. L. Jackson, Dissipative structure: An explanation and an ecological example, J. Theor. Biol., 37 (1972), 545–559. https://doi.org/10.1016/0022-5193(72)90090-2 doi: 10.1016/0022-5193(72)90090-2
|
| [27] |
F. Bauer, P. Horn, Y. Lin, G. Lippner, D. Mangoubi, S. T. Yau, Li-Yau inequality on graphs, J. Differ. Geom., 99 (2015), 359–405. https://doi.org/10.4310/jdg/1424880980 doi: 10.4310/jdg/1424880980
|
| [28] |
Y. Chung, Y. Lee, S. Chung, Extinction and positivity of the solutions of the heat equations with absorption on networks, J. Math. Anal. Appl., 380 (2011), 642–652. https://doi.org/10.1016/j.jmaa.2011.03.006 doi: 10.1016/j.jmaa.2011.03.006
|
| [29] |
A. Grigoryan, Y. Lin, Y. Yang, Yamabe type equations on graphs, J. Differ. Equations, 261 (2016), 4924–4943. https://doi.org/10.1016/j.jde.2016.07.011 doi: 10.1016/j.jde.2016.07.011
|
| [30] |
A. Grigoryan, Y. Lin, Y. Yang, Kazdan-Warner equation on graph, Calc. Var. Partial Differ. Equations, 55 (2016), 92. https://doi.org/10.1007/s00526-016-1042-3 doi: 10.1007/s00526-016-1042-3
|
| [31] |
M. Li, Z. Shuai, Global-stability problem for coupled systems of differential equations on networks, J. Differ. Equations, 248 (2010), 1–20. https://doi.org/10.1016/j.jde.2009.09.003 doi: 10.1016/j.jde.2009.09.003
|
| [32] |
H. Zhang, M. Small, X. Fu, G. Sun, B. Wang, Modeling the influence of information on the coevolution of contact networks and the dynamics of infectious diseases, Physica D, 241 (2012), 1512–1517. https://doi.org/10.1016/j.physd.2012.05.011 doi: 10.1016/j.physd.2012.05.011
|
| [33] |
C. Tian, S. Ruan, Pattern formation and synchronism in an allelopathic plankton model with delay in a network, SIAM J. Appl. Dyn. Syst., 18 (2019), 531–557. https://doi.org/10.1137/18M1204966 doi: 10.1137/18M1204966
|
| [34] |
Z. Liu, J. Chen, C. Tian, Blow-up in a network mutualistic model, Appl. Math. Lett., 106 (2020), 106402. https://doi.org/10.1016/j.aml.2020.106402 doi: 10.1016/j.aml.2020.106402
|
| [35] |
W. Gan, P. Zhu, Z. Liu, C. Tian, Delay-driven instability and ecological control in a food-limited population networked system, Nonlinear Dyn., 100 (2020), 4031–4044. https://doi.org/10.1007/s11071-020-05729-w doi: 10.1007/s11071-020-05729-w
|
| [36] |
L. Chang, M. Duan, G. Sun, Z. Jin, Cross-diffusion-induced patterns in an SIR epidemic model on complex networks, Chaos, 30 (2020), 013147. https://doi.org/10.1063/1.5135069 doi: 10.1063/1.5135069
|
| [37] |
C. Tian, Q. Zhang, L. Zhang, Global stability in a networked SIR epidemic model, Appl. Math. Lett., 107 (2020), 106444. https://doi.org/10.1016/j.aml.2020.106444 doi: 10.1016/j.aml.2020.106444
|
| [38] | H. Smith, An Introduction to Delay Differential Equations with Applications to the Life Sciences, Springer, 2011. https://doi.org/10.1007/978-1-4419-7646-8 |
| [39] |
Z. Liu, C. Tian, A weighed networked SIRS epidemic model, J. Differ. Equations, 269 (2020), 10995–11019. https://doi.org/10.1016/j.jde.2020.07.038 doi: 10.1016/j.jde.2020.07.038
|
| [40] | L. Pontryagin, V. Boltyanskii, R. Gamkrelize, E. Mishchenoko, The Mathematical Theory of Optimal Processes, Wiley, 1962. |
| [41] | W. Fleming, R. Rishel, Deterministic and Stochastic Optimal Control, Springer-Verlag, 1975. https://doi.org/10.1007/978-1-4612-6380-7 |




Figures(5)

Xiaomei Bao, Canrong Tian. Turing patterns in a networked vegetation model[J]. Mathematical Biosciences and Engineering, 2024, 21(11): 7601-7620. doi: 10.3934/mbe.2024334







 DownLoad:
DownLoad: