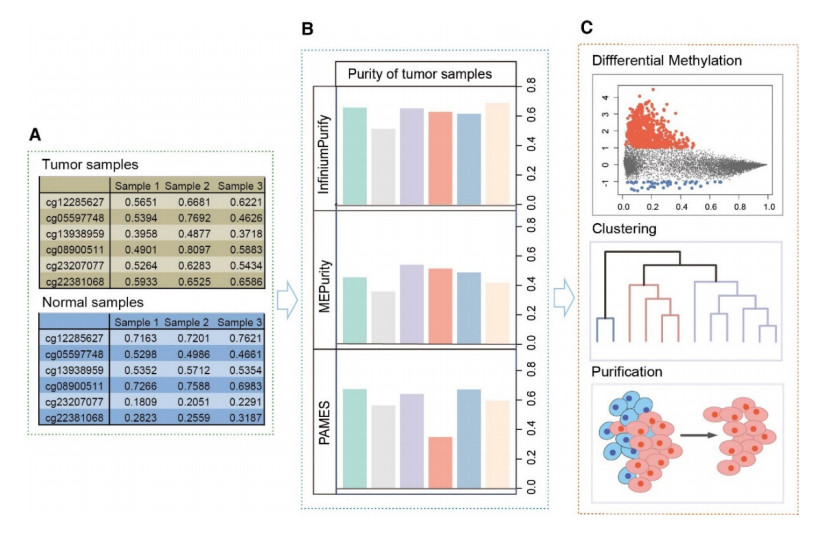
Proportion of cancerous cells in a tumor sample, known as "tumor purity", is a major source of confounding factor in cancer data analyses. Lots of computational methods are available for estimating tumor purity from different types of genomics data or based on different platforms, which makes it difficult to compare and integrate the estimated results. To rectify the deviation caused by tumor purity effect, a number of methods for downstream data analysis have been developed, including tumor sample clustering, association study and differential methylation between tumor samples. However, using these computational tools remains a daunting task for many researchers since they require non-trivial computational skills. To this end, we present Purimeth, an integrated web-based tool for estimating and accounting for tumor purity in cancer DNA methylation studies. Purimeth implements three state-of-the-art methods for tumor purity estimation from DNA methylation array data: InfiniumPurify, MEpurity and PAMES. It also provides graphical interface for various analyses including differential methylation (DM), sample clustering, and purification of tumor methylomes, all with the consideration of tumor purities. In addition, Purimeth catalogs estimated tumor purities for TCGA samples from nine methods for users to visualize and explore. In conclusion, Purimeth provides an easy-operated way for researchers to explore tumor purity and implement cancer methylation data analysis. It is developed using Shiny (Version 1.6.0) and freely available at http://purimeth.comp-epi.com/.
Citation: Nana Wei, Hanwen Zhu, Chun Li, Xiaoqi Zheng. Purimeth: an integrated web-based tool for estimating and accounting for tumor purity in cancer DNA methylation studies[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 8951-8961. doi: 10.3934/mbe.2021441
Proportion of cancerous cells in a tumor sample, known as "tumor purity", is a major source of confounding factor in cancer data analyses. Lots of computational methods are available for estimating tumor purity from different types of genomics data or based on different platforms, which makes it difficult to compare and integrate the estimated results. To rectify the deviation caused by tumor purity effect, a number of methods for downstream data analysis have been developed, including tumor sample clustering, association study and differential methylation between tumor samples. However, using these computational tools remains a daunting task for many researchers since they require non-trivial computational skills. To this end, we present Purimeth, an integrated web-based tool for estimating and accounting for tumor purity in cancer DNA methylation studies. Purimeth implements three state-of-the-art methods for tumor purity estimation from DNA methylation array data: InfiniumPurify, MEpurity and PAMES. It also provides graphical interface for various analyses including differential methylation (DM), sample clustering, and purification of tumor methylomes, all with the consideration of tumor purities. In addition, Purimeth catalogs estimated tumor purities for TCGA samples from nine methods for users to visualize and explore. In conclusion, Purimeth provides an easy-operated way for researchers to explore tumor purity and implement cancer methylation data analysis. It is developed using Shiny (Version 1.6.0) and freely available at http://purimeth.comp-epi.com/.

| [1] |
D. Aran, M. Sirota, A. J. Butte, Systematic pan-cancer analysis of tumour purity, Nat. Commun., 6 (2015), 8971. doi: 10.1038/ncomms9971

|
| [2] |
J. K. Rhee, Y. C. Jung, K. R. Kim, J. Yoo, J. Kim, Y. J. Lee, et al., Impact of tumor purity on immune gene expression and clustering analyses across multiple cancer types, Cancer Immunol. Res., 6 (2018), 87-97. doi: 10.1158/2326-6066.CIR-17-0201
|
| [3] |
A. E. Jaffe, R. A. Irizarry, Accounting for cellular heterogeneity is critical in epigenome-wide association studies, Genome Biol., 15 (2014), 1-9. doi: 10.1186/gb-2014-15-1-r1
|
| [4] |
S. L. Carter, K. Cibulskis, E. Helman, A. McKenna, H. Shen, T. Zack, et al., Absolute quantification of somatic DNA alterations in human cancer, Nat. Biotechnol., 30 (2012), 413-421. doi: 10.1038/nbt.2203
|
| [5] |
K. Yoshihara, M. Shahmoradgoli, E. Martínez, R. Vegesna, H. Kim, W. Torres-Garcia, et al., Inferring tumour purity and stromal and immune cell admixture from expression data, Nat. Commun., 4 (2013), 2612. doi: 10.1038/ncomms3612
|
| [6] |
M. Benelli, D. Romagnoli, F. Demichelis, Tumor purity quantification by clonal DNA methylation signatures, Bioinformatics, 34 (2018), 1642-1649. doi: 10.1093/bioinformatics/bty011
|
| [7] |
X. Zheng, N. Zhang, H. J. Wu, H. Wu, Estimating and accounting for tumor purity in the analysis of DNA methylation data from cancer studies, Genome Biol., 18 (2017), 1-14. doi: 10.1186/s13059-016-1139-1
|
| [8] | B. W. Liu, X. F. Yang, T. J. Wang, J. D. Lin, Y. Y. Kang, P. Jia, et al., MEpurity: estimating tumor purity using DNA methylation data, Bioinformatics, 35 (2019), 5298-5300. |
| [9] | S. Haider, S. Tyekucheva, D. Prandi, N. S. Fox, J. Ahn, A. W. Xu, et al., Systematic assessment of tumor purity and its clinical implications, JCO Precis. Oncol., 4 (2020), 995-1005. |
| [10] | F. Wang, N. Zhang, J. Wang, H. Wu, X. Zheng, Tumor purity and differential methylation in cancer epigenomics, Briefings Funct. Genomics, 15 (2016), 408-419. |
| [11] |
A. Paziewska, M. Dabrowska, K. Goryca, A. Antoniewicz, J. Dobruch, M. Mikula, et al., DNA methylation status is more reliable than gene expression at detecting cancer in prostate biopsy, Br. J. Cancer, 111 (2014), 781-789. doi: 10.1038/bjc.2014.337
|
| [12] |
N. Zhang, H. J. Wu, W. Zhang, J. Wang, H. Wu, X. Zheng, Predicting tumor purity from methylation microarray data, Bioinformatics, 31 (2015), 3401-3405. doi: 10.1093/bioinformatics/btv370
|
| [13] |
M. T. Gyparaki, E. K. Basdra, A. G. Papavassiliou, DNA methylation biomarkers as diagnostic and prognostic tools in colorectal cancer, J. Mol. Med., 91 (2013), 1249-1256. doi: 10.1007/s00109-013-1088-z
|
| [14] |
B. N. Lasseigne, T. C. Burwell, M. A. Patil, D. M. Absher, J. D. Brooks, R. M. Myers, DNA methylation profiling reveals novel diagnostic biomarkers in renal cell carcinoma, BMC Med., 12 (2014), 235. doi: 10.1186/s12916-014-0235-x
|
| [15] |
G. Nikolaidis, O. Y. Raji, S. Markopoulou, J. R. Gosney, J. Bryan, C. Warburton, et al., DNA methylation biomarkers offer improved diagnostic efficiency in lung cancer, Cancer Res., 72 (2012), 5692-5701. doi: 10.1158/0008-5472.CAN-12-2309
|
| [16] |
A. Farooq, S. Grønmyr, O. Ali, T. Rognes, K. Scheffler, M. Bjørås, et al., HMST-Seq-Analyzer: A new python tool for differential methylation and hydroxymethylation analysis in various DNA methylation sequencing data, Comput. Struct. Biotechnol. J., 18 (2020), 2877-2889. doi: 10.1016/j.csbj.2020.09.038
|
| [17] |
W. Zhang, Z. Li, N. Wei, H. J. Wu, X. Zheng, Detection of differentially methylated CpG sites between tumor samples with uneven tumor purities, Bioinformatics, 36 (2020), 2017-2024. doi: 10.1093/bioinformatics/btz885
|
| [18] |
W. Zhang, H. Feng, H. Wu, X. Zheng, Accounting for tumor purity improves cancer subtype classification from DNA methylation data, Bioinformatics, 33 (2017), 2651-2657. doi: 10.1093/bioinformatics/btx303
|





Figures(6)

Nana Wei, Hanwen Zhu, Chun Li, Xiaoqi Zheng. Purimeth: an integrated web-based tool for estimating and accounting for tumor purity in cancer DNA methylation studies[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 8951-8961. doi: 10.3934/mbe.2021441







 DownLoad:
DownLoad: