Citation: Lingling Li, Congbo Li, Li Li, Ying Tang, Qingshan Yang. An integrated approach for remanufacturing job shop scheduling with routing alternatives[J]. Mathematical Biosciences and Engineering, 2019, 16(4): 2063-2085. doi: 10.3934/mbe.2019101

| [1] | G. D. Li, M. Reimann and W. H. Zhang, When remanufacturing meets product quality improvement: The impact of production cost, Eur. J. Oper. Res., 271 (2018), 913–925. |
| [2] | P. V. Loon and L. N. V. Wassenhove, Assessing the economic and environmental impact of remanufacturing: a decision support tool for OEM suppliers, Int. J. Prod. Res., 56 (2017), 1662–1674. |
| [3] | M. Matsumoto, K. Chinen and H. Endo, Remanufactured auto parts market in Japan: Historical review and factors affecting green purchasing behavior, J. Clean Prod., 172 (2018), 4494–4505. |
| [4] | B. M. Liu, D. J. Chen and W. J. Zhou, The effect of remanufacturing and direct reuse on resource productivity of China's automotive production, J. Clean Prod., 194 (2018), 309–317. |
| [5] | Y. F. Zhang, S. C. Liu and Y. Liu, The 'Internet of Things' enabled real-time scheduling for remanufacturing of automobile engines, J. Clean Prod., 185 (2018), 562–575. |
| [6] | J. Zhou and Q. W. Deng, An environmental benefits and costs assessment model for remanufacturing process under quality uncertainty, J. Clean Prod., 186 (2018), 180–190. |
| [7] | R. Kumar and P. Ramachandran, Revenue management in remanufacturing: perspectives, review of current literature and research directions, Int. J. Prod. Res., 54 (2016), 2185–2201. |
| [8] | V. D. R. Guide, R. Srivastava and M. E. Kraus, Priority scheduling policies for repair shops, Int. J. Prod. Res., 38 (2000), 929–950. |
| [9] | V. D. R. Guide, G. C. Souza and E. V. D. Lann, Performance of static priority rules for shared facilities in a remanufacturing shop with disassembly and reassembly, Eur. J. Oper. Res., 164 (2005), 341–353. |
| [10] | R. H. Teunter, K. Kaparis and O. Tang, Multi-product economic lot scheduling problem with separate production lines for manufacturing and remanufacturing, Eur. J. Oper. Res., 191 (2008), 1241–1253. |
| [11] | S. Zanoni, A. Segerstedt, O. Tang, et al., Multi-product economic lot scheduling problem with manufacturing and remanufacturing using a basic period policy, Comput. Ind. Eng., 62 (2012), 1025–1033. |
| [12] | H. Sun, W. D. Chen, B. Y. Liu, et al., Economic lot scheduling problem in a remanufacturing system with returns at different quality grades, J. Clean Prod., 170 (2018), 559–569. |
| [13] | M. G. Kim, J. M. Yu and D. H. Lee, Solution algorithms for scheduling flow-shop-type remanufacturing systems. The 14th Asia Pacific Industrial Engineering and Management System Conference Vietnam, December 8–11, (2013). |
| [14] | P. B. Luh, D. Q. Yu, S. Soorapanth, et al., Relaxation based approach to schedule asset overhaul and repair services, IEEE T. Autom. Sci. Eng., 2 (2005), 145–157. |
| [15] | H. J. Wen, S. W. Hou, Z. H. Liu, et al., Integrated lot sizing and energy-efficient job shop scheduling problem in manufacturing/remanufacturing systems, Chaos Solitons Fractals, 105 (2017), 69–76. |
| [16] | R. Zhang, S. K. Ong and A. Y. C. Nee, A simulation-based genetic algorithm approach for remanufacturing process planning and scheduling, Appl. Soft. Comput., 37 (2016), 521–532. |
| [17] | C. B. Li, Y. Tang, C. C. Li, et al., A modeling approach to analyze variability of remanufacturing process routing, IEEE T. Autom. Sci. Eng., 10 (2013), 86–89. |
| [18] | S. E. Zhao, Y. L. Li and R. Fu, Fuzzy reasoning Petri nets and its application to disassembly sequence decision-making for the end-of-life product recycling and remanufacturing, Int. J. Comput. Integr. Manuf., 27 (2014), 415–421. |
| [19] | Y. Tang, M. C. Zhou and M. M Gao, Fuzzy-Petri-net-based disassembly planning considering human factors, IEEE T. Syst. Man Cybern. Syst., 36 (2006), 718–726. |
| [20] | L. L. Li, C. B. Li and Y. Tang, An integrated approach of reverse engineering aided remanufacturing process for worn components, Robot. Comput. Integr. Manuf., 48 (2017), 39–50. |
| [21] | D. H. Wu and W. Zheng, Formal model-based quantitative safety analysis using timed Coloured Petri Nets, Reliab. Eng. Syst. Saf., 176 (2018), 62–79. |
| [22] | H. L. Liao, Q. W. Deng and Y. R. Wang, An environmental benefits and costs assessment model for remanufacturing process under quality uncertainty, J. Clean Prod., 178 (2018), 45–58. |
| [23] | G. D. Li, M. Reimann and W. H. Zhang, When remanufacturing meets product quality improvement: The impact of production cost, Eur. J. Oper. Res., 271 (2018), 913–925. |
| [24] | J. Y. Sheng and D. Prescott, A hierarchical coloured Petri net model of fleet maintenance with cannibalisation, Reliab. Eng. Syst. Saf., 168 (2017), 290–305. |
| [25] | S. A. Hussain, N. A. Khan and A. Sadiq, Simulation, modeling and analysis of master node election algorithm based on signal strength for VANETs through Colored Petri nets, Neural Comput. Appl., 29 (2018), 1243–1259. |
| [26] | Y. W. Si, V. I. Chan, M. Dumas, et al., A Petri nets based generic genetic algorithm framework for resource optimization in business processes, Simul. Model. Pract. Theory, 86 (2018), 72–101. |
| [27] | A. Assad and K. Deep, A Hybrid Harmony search and Simulated Annealing algorithm for continuous optimization, Inf. Sci., 450 (2018), 246–266. |
| [28] | Z. Y. Liu, Z. S. Liu, Z. P. Zhu, et al., Simulated annealing for a multi-level nurse rostering problem in hemodialysis service, Appl. Soft. Comput., 64 (2017), 148–160. |
| [29] | F. Erchiqui, Application of genetic and simulated annealing algorithms for optimization of infrared heating stage in thermoforming process, Appl. Therm. Eng., 128 (2018), 1273–1272. |
| [30] | X. Y. Li, C. Lu, L. Gao, et al., An effective multi-objective algorithm for energy efficient scheduling in a real-life welding shop, IEEE T. Ind. Inform., 14 (2018), 5400–5409. |
| [31] | S. Hore, A. Chatterjee and A. Dewanji, Improving variable neighborhood search to solve the traveling salesman problem, Appl. Soft. Comput., 68 (2018), 83–91. |
| [32] | X. Y. Li, L. Gao, Q. K. Pan, et al., An effective hybrid genetic algorithm and variable neighborhood search for integrated process planning and scheduling in a packaging machine workshop, IEEE T. Syst. Man. Cybern. Syst., (2018). |
| [33] | Y. Z. Zhou, W. C. Yi, L. Gao, et al., Adaptive differential evolution with sorting crossover rate for continuous optimization problems, IEEE T. Cybern., 47 (2017), 2742–2753. |
| [34] | X. Y. Li and L. Gao, An effective hybrid genetic algorithm and tabu search for flexible job shop scheduling problem. Int. J. Prod. Econ., 174 (2016), 93–110. |
| [35] | G. R. Amin and A. El-Bouri, A minimax linear programming model for dispatching rule selection, Comput. Ind. Eng., 121 (2018), 27–35. |
| [36] | P. Neammanee and M. Reodecha, A memetic algorithm-based heuristic for a scheduling problem in printed circuit board assembly, Comput. Ind. Eng., 56 (2009), 294–305. |
| [37] | B. N. Silva, M. Khan and K. Han, Load balancing integrated least slack time-based appliance scheduling for smart home energy management, Sensors, 18 (2018). |
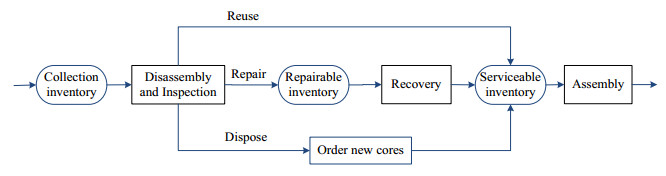
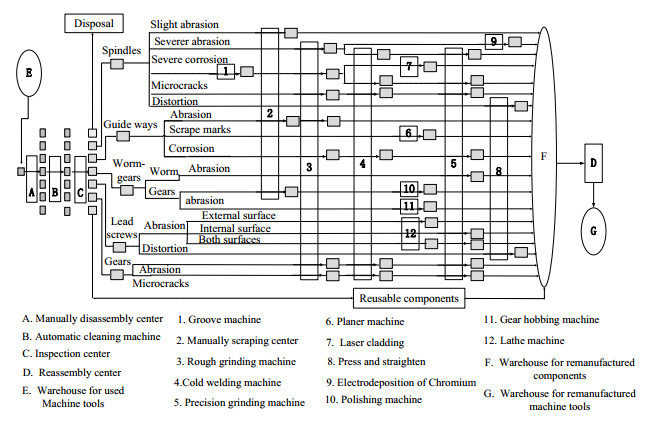
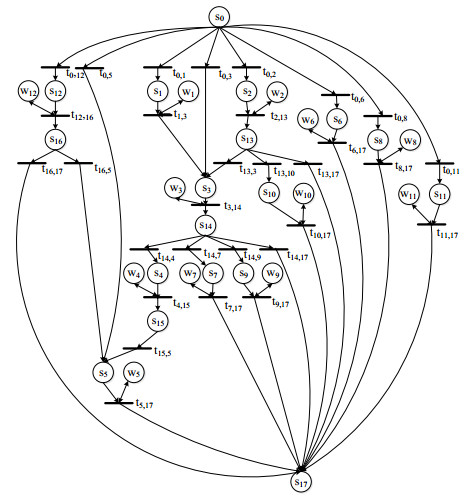
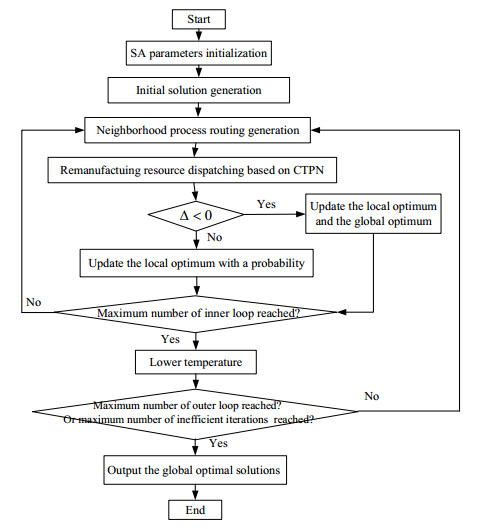

Figures(6) / Tables(8)

Lingling Li, Congbo Li, Li Li, Ying Tang, Qingshan Yang. An integrated approach for remanufacturing job shop scheduling with routing alternatives[J]. Mathematical Biosciences and Engineering, 2019, 16(4): 2063-2085. doi: 10.3934/mbe.2019101







 DownLoad:
DownLoad: