Citation: Xu Zhang, Dongdong Chen, Wenmin Yang, JianhongWu. Identifying candidate diagnostic markers for tuberculosis: A critical role of co-expression and pathway analysis[J]. Mathematical Biosciences and Engineering, 2019, 16(2): 541-552. doi: 10.3934/mbe.2019026

| [1] | A. Zumla, A. George, V. Sharma and R.H. Herbert, Baroness Masham of Ilton, Oxley A, Oliver M, The who 2014 global tuberculosis report-further to go, Lancet Glob. Health, 3 (2015), 10–12. |
| [2] | WHO Representative Office, China-Tuberculosis in China 2015. Available from: http://www. wpro.who.int/china/mediacentre/factsheets/tuberculosis/en/. |
| [3] | A. E. Gorna, R. P. Bowater and J. Dziadek, DNA repair systems and the pathogenesis of Mycobacterium tuberculosis: varying activities at different stages of infection, Clin. Sci., 119 (2010), 187–202. |
| [4] | B. J. Marais, K. Lönnroth, S. D. Lawn, G. B. Migliori, P. Mwaba, P. Glaziou, P. Glaziou, M. Bates, R. Colagiuri, L. Zijenah, S. Swaminathan, Z.A. Memish, M. Pletschette, M. Hoelscher, I. Abubakar, R. Hasan, A. Zafar, G. Pantaleo, G. Craig, P. Kim, M. Maeurer, M. Schito and A. Zumla, Tuberculosis comorbidity with communicable and non-communicable diseases: integrating health services and control efforts, Lancet Infect. Dis., 13 (2013), 436–448. |
| [5] | D. Snchez, M. Rojas, I. Hernndez, D. Radzioch, L.F. Garca and L.F. Barrera, Role of TLR2- and TLR4-mediated signaling in Mycobacterium tuberculosis -induced macrophage death, Cell. Immunol., 260 (2010), 128–136. |
| [6] | J. Maertzdorf, D. Repsilber, S.K. Parida, K. Stanley, T. Roberts, G. Black, G. Walzl and S.H. Kaufmann, Human gene expression profiles of susceptibility and resistance in tuberculosis, Gene. Immu., 12 (2011), 15–22. |
| [7] | G. K. Smyth, Linear models and empirical bayes methods for assessing differential expression in microarray experiments, Statist. Appl. Genet. Molecul. Biol., 3 (2004), 1544–6115. |
| [8] | D. J. Mccarthy and G. K. Smyth,Testing significance relative to a fold-change threshold is a TREAT, Bioinformatics, 25 (2009), 765–771. |
| [9] | M. Platten, N. von Knebel Doeberitz, I. Oezen, W. Wick and K. Ochs, Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors, Front. Immunol., 5 (2014), 1–7. |
| [10] | F. Li, R. Zhang, S. Li and J. IDOL. Liu, An important immunotherapy target in cancer treatment, Int. Immunopharmacol., 47 (2017), 70–77. |
| [11] | I. N. Maria, C. E. Steenwijk and S. A. Ijpma,Contrasting expression pattern of RNA-sensing receptors TLR7, RIG-I and MDA5 in interferon-positive and interferon-negative patients with primary Sjögren's syndrome, An. Rheumat. Dis., 76 (2016), 721–730. |
| [12] | A.R. Shenoy, D.A.Wellington, P. Kumar, H. Kassa, C.J. Booth, P. Cresswell and J.D. MacMicking, GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals, Science, 336 (2012), 481–485. |
| [13] | C. Krapp, D. Hotter, A. Gawanbacht, J. P. Mclaren, F. S. Kluge, M. C. Stürzel, K. Mack, E. Reith, S. Engelhart , A. Ciuff, V. Hornung, D. Sauter, A. Telenti, and F. Kirchhoff, Guanylate Binding Protein (GBP) 5 Is an Interferon-Inducible Inhibitor of HIV-1 Infectivity, Cell Host Microb., 19 (2016), 504–514. |
| [14] | E. T. Pronk,W. J. Veen, J. R. Vandebriel, V. H. Loveren, P. E. Vink and L. J. Pennings, Comparison of the molecular topologies of stress-activated transcription factors HSF1, AP-1, NRF2, and NF-kB in their induction kinetics of HMOX1, Biosystems, 124 (2014), 75–85. |
| [15] | K. Ramsauer, M. Farlik, G. Zupkovitz, C. Seiser and T. Decker, Distinct modes of action applied by transcription factors STAT1 and IRF1 to initiate transcription of the IFN- inducible gbp2 gene, Proceed. Nat. Aca. Sci. US Am., 104 (2007), 2849–2854. |
| [16] | M. Qi, M. Ge and L. Huang, Up-regulation of GBP2 is Associated with Neuronal Apoptosis in Rat Brain Cortex Following Traumatic Brain Injury, Neurochem. Res., 42 (2017), 1515–1523. |
| [17] | D. Hober and P. Sauter, Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host, Nat. Rev. Endocrinol., 6 (2010), 279–289. |
| [18] | H. Takedatsu, S. K. Michelsen, B. Wei, J. C. Landers, S. L. Thomas, D. Dhall, J. Braun and S.R. Targan, TL1A (TNFSF15) Regulates the Development of Chronic Colitis By Modulating both T helper (TH) 1 and TH17 Activation, Gastroenterology, 135 (2008), 552–567. |
| [19] | H. Deng, H. Liu, B. Zhai, K. Zhang, G. Xu, X. Peng, Q. Zhang and L. Li, Vascular endothelial growth factor suppressesTNFSF15 production in endothelial cells by stimulating miR-31 and miR- 20a expression via activation of Akt and Erk signals, Febs. Open Biol., 7 (2017), 108–117. |
| [20] | F. Mackay and P. Schneider , TACI, an enigmatic BAFF/APRIL receptor, with new unappreciated biochemical and biological properties, Cytok. Growth Factor Rev., 19 (2008), 263–276. |
| [21] | Y. E. APAMoon, H. J. Lee, W. J. Lee, H. J. Song and S. Pyo, ROS/Epac1-mediated Rap1/NF-$\kappa$B activation is required for the expression of BAFF in Raw264.7 murine macrophages, Cell. Signal., 23 (2011), 1479–1488. |
| [22] | H. Akca, A. Demiray, O. Tokgun and J. Yokota, Invasiveness and anchorage independent growth ability augmented by PTEN inactivation through the PI3K/AKT/NF-$\kappa$B pathway in lung cancer > cells, Lung Cancer, 73 (2011), 302–309. |
| [23] | M. Vucur, C. Roderburg, K. Bettermann, F. Tacke, M. Heikenwalder, C. Trautwein and T. Luedde, Mouse models of hepatocarcinogenesis: what can we learn for the prevention of human hepatocellular carcinoma?, Oncotarget, 1 (2010), 373–378. |
| [24] | H. P. Krammer, R. Arnold and I. N. Lavrik, Life and death in peripheral T cells, Nat. Rev. Immunol., 7 (2007), 532–542. |
| [25] | K. Ozato, M. D. Shin, H. T. Chang, Iii and H. C. M,TRIM family proteins and their emerging roles in innate immunity, Nat. Rev. Immunol., 8 (2008), 849–860. |
| [26] | Z. Sepehri, Z. Kiani, F. Javadian, A. N. Akbar, F. Kohan, S. Sepehrikia, S.S. Javan, H. Aali, H. Daneshvar and D. Kennedy, TLR3 and its roles in the pathogenesis of type 2 diabetes, Cell Mol. Biol. (Noisy-le-grand), 61 (2015), 46–50. |
| [27] | Z. Xia, G. Xu , X. Yang, N. Peng, Q. Zuo, S. Zhu, H. Hao, S. Liu and Y. Zhu, Inducible TAP1 Negatively Regulates the Antiviral Innate Immune Response by Targeting the TAK1 Complex, J. Immunol., 198 (2017), 3690–3704. |
| [28] | M. Trapecar, A. Goropevsek, M. Gorenjak, L. Gradisnik and S. M. Rupnik, A Co-Culture Model of the Developing Small Intestine Offers New Insight in the Early Immunomodulation of Enterocytes and Macrophages by Lactobacillus spp. through STAT1 and NF-$\kappa$B p65 Translocation, Plos One, 9 (2014), 1–8. |
| [29] | M. Mondini, S. Costa, S. Sponza, F. Gugliesi, M. Gariglio and S. Landolfo, The interferoninducible HIN-200 gene family in apoptosis and inflammation: implication for autoimmunity, Autoimmunity, 43 (2010), 226–231. |
| [30] | A. Maji, R. Misra, K. A. Mondal, D. Kumar, D. Bajaj, A. Singhal, G. Arora, A. Bhaduri, A. Sajid, S. Bhatia, S. Singh, H. Singh, V. Rao, D. Dash, S.E. Baby, M.J. Sarojini, A. Chaudhary, R.S. Gokhale and Y. Singh, Expression profiling of lymph nodes in tuberculosis patients reveal inflammatory milieu at site of infection, Sci. Rep., 5 (2015), 1–10. |
| [31] | M. Bawadekar, M. A. De, I. C. Lo, G. Baldanzi, V. Caneparo, A. Graziani, S. Landolfo and M. Gariglio, The Extracellular IFI16 Protein Propagates Inflammation in Endothelial Cells Via p38 MAPK and NF-$\kappa$B p65 Activation, J. Interferon. Cytokine. Res., 35 (2015), 441–453. |
| [32] | R. N. Han, A. H. Oh, Y. S. Nam, D. P. Moon,W. D. Kim, M. H. Kim, and H.J. Jeong, TSLP Induces Mast Cell Development and Aggravates Allergic Reactions through the Activation of MDM2 and STAT6, J. Invest. Dermatol., 134 (2014), 2521–2530. |
| [33] | K. Yao, Q. Chen, Y. Wu, F. Liu, X. Chen and Y. Zhang, Unphosphorylated STAT1 represses apoptosis in macrophages during Mycobacterium tuberculosis infection, J. Cell Sci., 130 (2017), 1740–1751. |
| [34] | Z. Cheng, Y. Yi, S. Xie, H. Yu, H. Peng and G. Zhang, The effect of the JAK2 inhibitor TG101209 against T cell acute lymphoblastic leukemia (T-ALL) is mediated by inhibition of JAK-STAT signaling and activation of the crosstalk between apoptosis and autophagy signaling, Oncotarget, 8 (2017), 106753–106763. |
| [35] | W. He, Q. Wang, J. Xu, X. Xu, MT. Padilla, G. Ren, X. Gou and Y. Lin, Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation, Autophagy, 8 (2012), 1811–1821. |
| [36] | M. Yang, L. Liu, M. Xie, X. Sun, Y. Yu, R. Kang, L. Yang, S. Zhu, L. Cao and D. Tang, Poly-ADPribosylation of HMGB1 regulates TNFSF10/TRAIL resistance through autophagy, Autophagy, 11 (2015), 214–224. |
| [37] | L.X. Xu, S. Grimaldo, J.W. Qi, G.L. Yang, T.T. Qin, H.Y. Xiao, R. Xiang, Z. Xiao, L.Y. Li and Z.S. Zhang, Death receptor 3 mediates TNFSF15- and TNF-induced endothelial cell apoptosis, Int. J. Biochem. Cell Biol., 55 (2014), 109–118. |
| [38] | A. Shrivastava, VEGI, a new member of the TNF family activates Nuclear Factor-$\kappa$B and c-Jun N-terminal kinase and modulates cell growth, Oncogene, 18 (1999), 6496–6504. |
| [39] | Y. Piao, C. Thomas, L. Holmes, V. Henry and D. J. Groot, ET-45KNOCKDOWN of TNFSF13B induces glioma stem cell apoptosis, Neuro-Oncology, 16 (2014), 79–95. |
| [40] | R. Rosell, T. G. Bivona and N. Karachaliou, Genetics and biomarkers in personalisation of lung cancer treatment, Lancet, 382 (2013), 720–731. |
| [41] | U. Gaur and B. B. Aggarwal, Regulation of proliferation, survival and apoptosis by members of the TNF superfamily, Biochem. Pharmacol., 66 (2003), 1403–1408. |
| [42] | X. Zhao, X. Liu and S. Ling, Parthenolide induces apoptosis via TNFRSF10B and PMAIP1 pathways in human lung cancer cells, J. Experiment. Clin. Cancer Res., 33 (2014), 1–11. |
| [43] | F.H. Wu, Y. Yuan, D. Li, S.J. Liao, B. Yan, J.J. Wei, Y.H. Zhou, J.H. Zhu, G.M. Zhang and Z.H. Feng, Extracellular HSPA1A promotes the growth of hepatocarcinoma by augmenting tumor cell proliferation and apoptosis-resistance, Cancer Lett., 317 (2012), 157–164. |
| [44] | F.H. Wu, Y. Yuan, D. Li, S.J. Liao, B. Yan, J.J. Wei, Y.H. Zhou, J.H. Zhu, G.M. Zhang and Z.H. Feng,Mycobacterium tuberculosis PE13 (Rv1195) manipulates the host cell fate via p38-ERK-NF- $\kappa$B axis and apoptosis, Apoptosis, 21 (2016), 795–808. |
| [45] | X. Yu, C. Li,W. Hong,W. Pan and J. Xie, Autophagy during Mycobacterium tuberculosis infection and implications for future tuberculosis medications, Cellul. Signal., 25 (2013), 1272–1278. |
| [46] | D.M. Shin, B.Y. Jeon, H.M. Lee, H.S. Jin, J.M. Yuk, C.H. Song, S.H. Lee, Z.W. Lee, S.N. Cho, J.M. Kim, R.L. Friedman and E.K. Jo, Mycobacterium tuberculosis Eis Regulates Autophagy, Inflammation, and Cell Death through Redox-dependent Signaling, Plos Pathogens, 6 (2010), 1–15. |
| [47] | J. Lee, G. H. Remold, H. M. Ieong and H. Kornfeld, Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosis is mediated by a novel caspase-independent pathway, J. Immunol., 176 (2006), 4267–4274. |
| [48] | M. J. Yuk and E. K. Jo, Host immune responses to mycobacterial antigens and their implications for the development of a vaccine to control tuberculosis, Clin. Experiment. Vac. Res., 3 (2014), 155–167. |
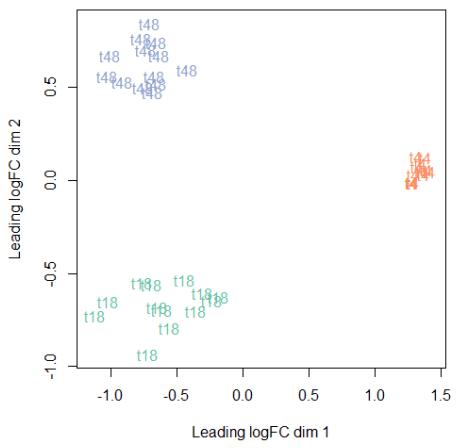
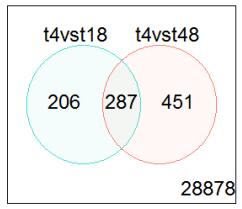
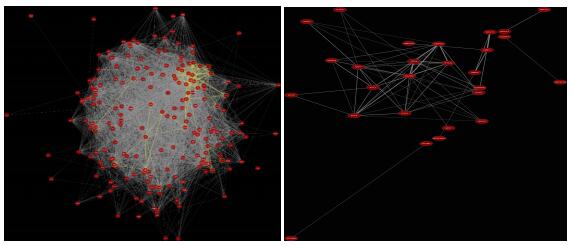
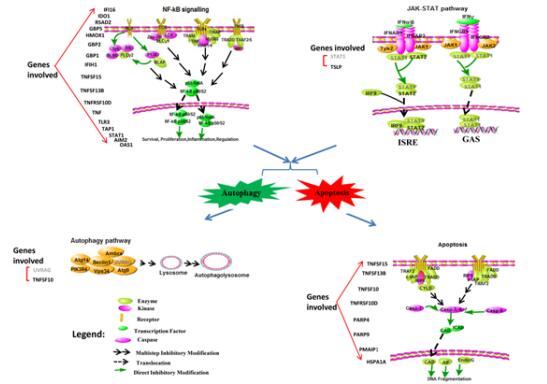
Figures(4) / Tables(3)

Xu Zhang, Dongdong Chen, Wenmin Yang, JianhongWu. Identifying candidate diagnostic markers for tuberculosis: A critical role of co-expression and pathway analysis[J]. Mathematical Biosciences and Engineering, 2019, 16(2): 541-552. doi: 10.3934/mbe.2019026







 DownLoad:
DownLoad: