Citation: Cicely K. Macnamara, Mark A. J. Chaplain. Spatio-temporal models of synthetic genetic oscillators[J]. Mathematical Biosciences and Engineering, 2017, 14(1): 249-262. doi: 10.3934/mbe.2017016

| [1] | [ S. Busenberg,J. M. Mahaffy, Interaction of spatial diffusion and delays in models of genetic control by repression, J. Math. Biol., 22 (1985): 313-333. |
| [2] | [ A. Cangiani,R. Natalini, A spatial model of cellular molecular trafficking including active transport along microtubules, J. Theor. Biol., 267 (2010): 614-625. |
| [3] | [ M. A. J. Chaplain,M. Ptashnyk,M. Sturrock, Hopf bifurcation in a gene regulatory network model: Molecular movement causes oscillations, Math. Models Method Appl. Sci., 25 (2015): 1179-1215. |
| [4] | [ Y. Y. Chen,K. E. Galloway,C. D. Smolke, Synthetic biology: Advancing biological frontiers by building synthetic systems, Genome Biology, 13 (2012): p240. |
| [5] | [ L. Dimitrio,J. Clairambault,R. Natalini, A spatial physiological model for p53 intracellular dynamics, J. Theor. Biol., 316 (2013): 9-24. |
| [6] | [ J. Eliaš,J. Clairambault, Reaction-diffusion systems for spatio-temporal intracellular protein networks: A beginner's guide with two examples, Comp. Struct. Biotech. J., 10 (2014): 14-22. |
| [7] | [ J. Eliaš,L. Dimitrio,J. Clairambault,R. Natalini, Modelling p53 dynamics in single cells: Physiologically based ODE and reaction-diffusion PDE models, Phys. Biol., 11 (2014): 045001. |
| [8] | [ J. Eliaš,L. Dimitrio,J. Clairambault,R. Natalini, The p53 protein and its molecular network: Modelling a missing link between DNA damage and cell fate, Biochim. Biophys. Acta (BBA Proteins and Proteomics), 1844 (2014): 232-247. |
| [9] | [ M. B. Elowitz,S. Leibler, A synthetic oscillatory network of transcriptional regulators, Nature, 403 (2000): 335-338. |
| [10] | [ L. Glass,S. A. Kauffman, Co-operative components, spatial localization and oscillatory cellular dynamics, J. Theor. Biol., 34 (1972): 219-237. |
| [11] | [ B. C. Goodwin, Oscillatory behaviour in enzymatic control processes, Adv. Enzyme Regul., 3 (1965): 425-428. |
| [12] | [ K. E. Gordon,I. M. M. V. Leeuwen,S. Lain,M. A. J. Chaplain, Spatio-temporal modelling of the p53-{M}dm2 oscillatory system, Math. Model. Nat. Phenom., 4 (2009): 97-116. |
| [13] | [ J. S. Griffith, Mathematics of cellular control processes. Ⅰ. negative feedback to one gene, J. Theor. Biol., 20 (1968): 202-208. |
| [14] | [ H. Hirata,S. Yoshiura,T. Ohtsuka,Y. Bessho,T. Harada,K. Yoshikawa,R. Kageyama, Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop, Science, 298 (2002): 840-843. |
| [15] | [ M. H. Jensen,J. Sneppen,G. Tiana, Sustained oscillations and time delays in gene expression of protein Hes1, FEBS Lett., 541 (2003): 176-177. |
| [16] | [ R. Kagemyama,T. Ohtsuka,T. Kobayashi, The Hes1 gene family: Repressors and oscillators that orchestrate embryogenesis, Development, 134 (2007): 1243-1251. |
| [17] | [ T. Kobayashi,R. Kageyama, Hes1 regulates embryonic stem cell differentiation by suppressing notch signaling, Genes to Cells, 15 (2010): 689-698. |
| [18] | [ T. Kobayashi,R. Kageyama, Hes1 oscillations contribute to heterogeneous differentiation responses in embryonic stem cells, Genes, 2 (2011): 219-228. |
| [19] | [ T. Kobayashi,H. Mizuno,I. Imayoshi,C. Furusawa,K. Shirahige,R. Kageyama, The cyclic gene Hes1 contributes to diverse differentiation responses of embryonic stem cells, Genes & Development, 23 (2009): 1870-1875. |
| [20] | [ G. Lahav,N. Rosenfeld,A. Sigal,N. Geva-Zatorsky,A. J. Levine,M. B. Elowitz,U. Alon, Dynamics of the p53-Mdm2 feedback loop in individual cells, Nature Genet., 36 (2004): 147-150. |
| [21] | [ E. Lieberman-Aiden,N. L. van Berkum,L. Williams,M. Imakaev,T. Ragoczy,A. Telling,I. Amit,B. R. Lajoie,P. J. Sabo,M. O. Dorschner,R. Sandstrom,B. Bernstein,M. A. Bender,M. Groudine,A. Gnirke,J. Stamatoyannopoulos,L. A. Mirny,E. S. Lander,J. Dekker, Comprehensive mapping of long range interactions reveals folding principles of the human genome, Science, 326 (2009): 289-293. |
| [22] | [ R.-T. Liu,S.-S. Liaw,P. K. Maini, Oscillatory turing patterns in a simple reaction-diffusion system, J. Korean Phys. Soc., 50 (2007): 234-238. |
| [23] | [ J. M. Mahaffy, Genetic control models with diffusion and delays, Math. Biosci., 90 (1988): 519-533. |
| [24] | [ J. M. Mahaffy,C. V. Pao, Models of genetic control by repression with time delays and spatial effects, J. Math. Biol., 20 (1984): 39-57. |
| [25] | [ R. Milo,S. Shen-Orr,S. Itzkovitz,N. Kashtan,D. Chklovskii,U. Alon, Network motifs: Simple building blocks of complex networks, Science, 298 (2002): 824-827. |
| [26] | [ H. Momiji,N. A. M. Monk, Dissecting the dynamics of the Hes1 genetic oscillator, J. Theor. Biol., 254 (2008): 784-798. |
| [27] | [ N. A. M. Monk, Oscillatory expression of Hes1, p53, and NF-κB driven by transcriptional time delays, Curr. Biol., 13 (2003): 1409-1413. |
| [28] | [ F. Naqib,T. Quail,L. Musa,H. Vulpe,J. Nadeau,J. Lei,L. Glass, Tunable oscillations and chaotic dynamics in systems with localized synthesis, Phys. Rev. E, 85 (2012): 046210. |
| [29] | [ E. L. O'Brien,E. Van Itallie,M. R. Bennett, Modeling synthetic gene oscillators, Math. Biosci., 236 (2012): 1-15. |
| [30] | [ O. Purcell,N. J. Savery,C. S. Grierson,M. di Bernardo, A comparative analysis of synthetic genetic oscillators, J. R. Soc. Interface, 7 (2010): 1503-1524. |
| [31] | [ L. Sang,H. A. Coller,M. J. Roberts, Control of the reversibility of cellular quiescence by the transcriptional repressor HES1, Science, 321 (2008): 1095-1100. |
| [32] | [ Y. Schaerli,A. Munteanu,M. Gili,J. Cotterell,J. Sharpe,M. Isalan, A unified design space of synthetic stripe-forming networks, Nat. Commun., 5 (2014): p4905. |
| [33] | [ R. M. Shymko,L. Glass, Spatial switching in chemical reactions with heterogeneous catalysis, J. Chem. Phys., 60 (1974): 835-841. |
| [34] | [ R. D. Skeel,M. Berzins, A method for the spatial discretization of parabolic equations in one space variable, SIAM J. Sci. and Stat. Comput., 11 (1990): 1-32. |
| [35] | [ M. Sturrock,A. Hellander,A. Matzavinos,M. A. J. Chaplain, Spatial stochastic modelling of the Hes1 gene regulatory network: Intrinsic noise can explain heterogeneity in embryonic stem cell differentiation, J. R. Soc. Interface, 10 (2013): 20120988. |
| [36] | [ M. Sturrock,A. J. Terry,D. P. Xirodimas,A. M. Thompson,M. A. J. Chaplain, Spatio-temporal modelling of the Hes1 and p53-Mdm2 intracellular signalling pathways, J. Theor. Biol., 273 (2011): 15-31. |
| [37] | [ M. Sturrock,A. J. Terry,D. P. Xirodimas,A. M. Thompson,M. A. J. Chaplain, Influence of the nuclear membrane, active transport and cell shape on the Hes1 and p53--Mdm2 pathways: Insights from spatio-temporal modelling, Bull. Math. Biol., 74 (2012): 1531-1579. |
| [38] | [ Z. Szymańska,M. Parisot,M. Lachowicz, Mathematical modeling of the intracellular protein dynamics: The importance of active transport along microtubules, J. Theor. Biol., 363 (2014): 118-128. |
| [39] | [ G. Tiana,M. H. Jensen,K. Sneppen, Time delay as a key to apoptosis induction in the p53 network, Eur. Phys. J., 29 (2002): 135-140. |
| [40] | [ B. Yordanov,N. Dalchau,P. K. Grant,M. Pedersen,S. Emmott,J. Haseloff,A. Phillips, A computational method for automated characterization of genetic components, ACS Synthetic Biology, 3 (2014): 578-588. |
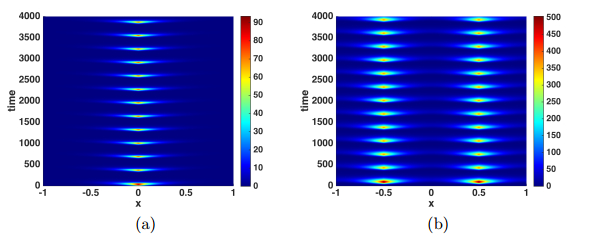
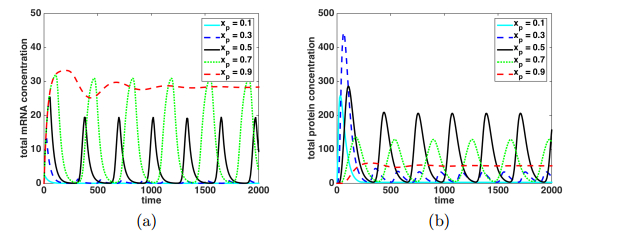
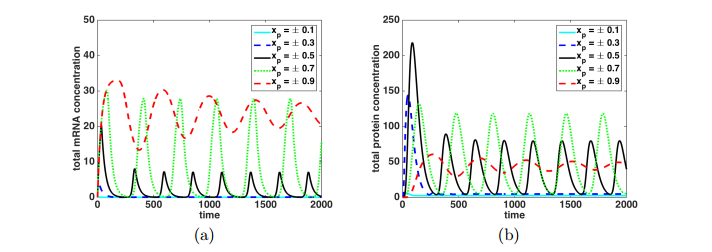
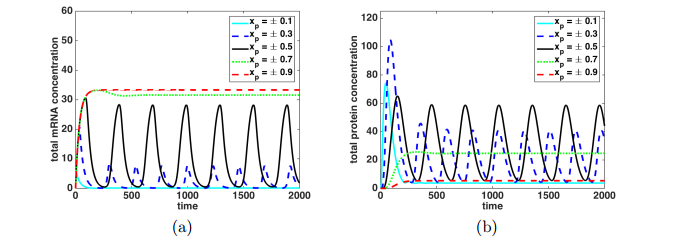
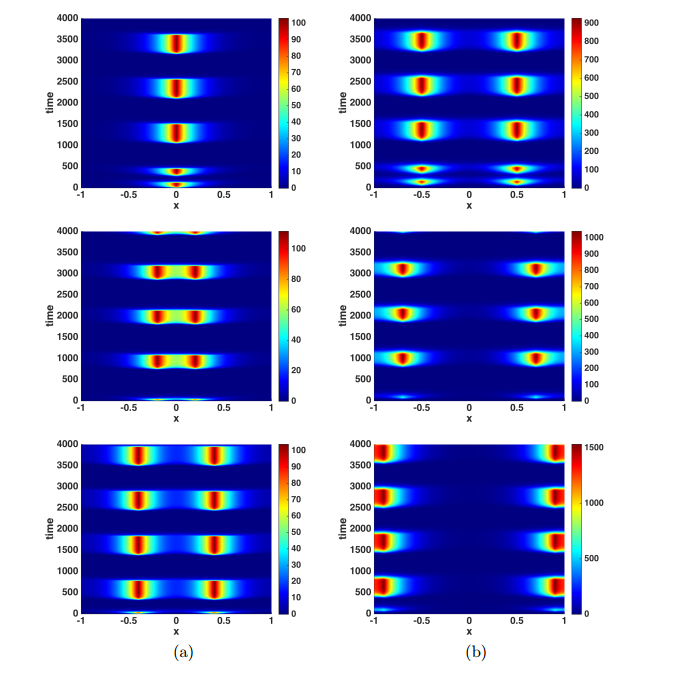
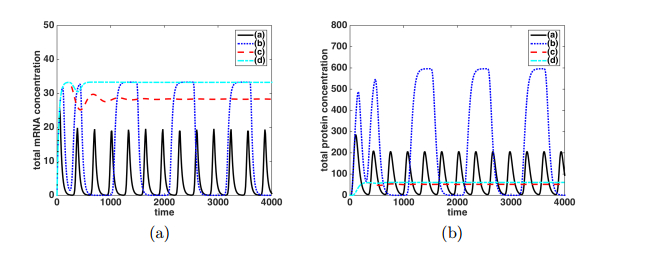
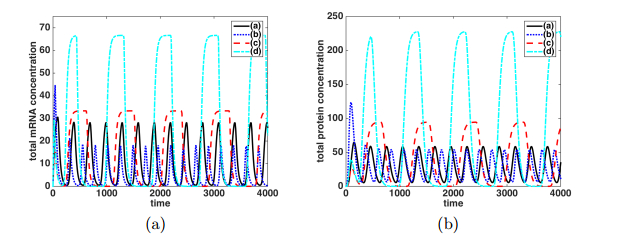
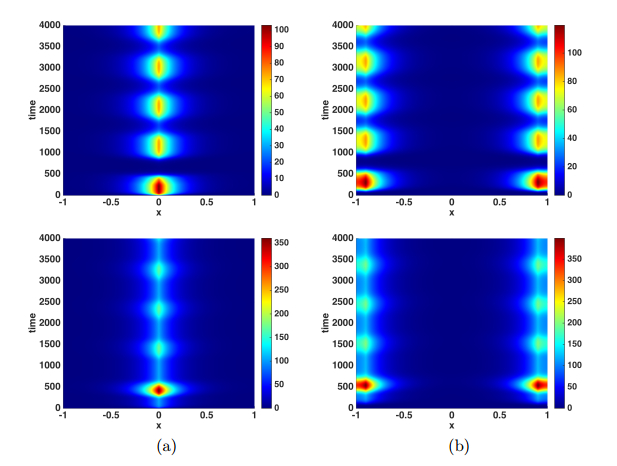
Figures(9)

Cicely K. Macnamara, Mark A. J. Chaplain. Spatio-temporal models of synthetic genetic oscillators[J]. Mathematical Biosciences and Engineering, 2017, 14(1): 249-262. doi: 10.3934/mbe.2017016







 DownLoad:
DownLoad: