Citation: Morgan Robinson, Brenda Yasie Lee, Zoya Leonenko. Drugs and drug delivery systems targeting amyloid-β in Alzheimer's disease[J]. AIMS Molecular Science, 2015, 2(3): 332-358. doi: 10.3934/molsci.2015.3.332

| [1] | Prince M, Jackson J (2009) Alzheimer's Disease International World Alzheimer Report 2009. London. 1-96 p. |
| [2] |
Takeda A, Loveman E, Clegg A, et al. (2006) A systematic review of the clinical effectiveness of donepezil, rivastigmine and galantamine on cognition, quality of life and adverse events in Alzheimer's disease. Int J Geriatr Psychiatry 21: 17-28. doi: 10.1002/gps.1402

|
| [3] |
Ong KT, Villemagne VL, Bahar-Fuchs A, et al. (2015) Abeta imaging with 18F-florbetaben in prodromal Alzheimer's disease: a prospective outcome study. J Neurol Neurosurg Psychiatry 86: 431-436. doi: 10.1136/jnnp-2014-308094
|
| [4] |
Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362: 329-344. doi: 10.1056/NEJMra0909142
|
| [5] |
Sperling RA, Aisen PS, Beckett LA, et al. (2011) Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7: 280-292. doi: 10.1016/j.jalz.2011.03.003
|
| [6] |
Petkova AT, Ishii Y, Balbach JJ, et al. (2002) A structural model for Alzheimer's beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci U S A 99: 16742-16747. doi: 10.1073/pnas.262663499
|
| [7] | Matsuzaki K (2014) How do membranes initiate Alzheimer's Disease? Formation of toxic amyloid fibrils by the amyloid beta-protein on ganglioside clusters. Acc Chem Res 47: 2397-2404. |
| [8] | Cecchi C, Stefani M (2013) The amyloid-cell membrane system. The interplay between the biophysical features of oligomers/fibrils and cell membrane defines amyloid toxicity. Biophys Chem 182: 30-43. |
| [9] |
Drolle E, Gaikwad RM, Leonenko Z (2012) Nanoscale electrostatic domains in cholesterol-laden lipid membranes create a target for amyloid binding. Biophys J 103: L27-29. doi: 10.1016/j.bpj.2012.06.053
|
| [10] |
Drolle E, Hane F, Lee B, et al. (2014) Atomic force microscopy to study molecular mechanisms of amyloid fibril formation and toxicity in Alzheimer's disease. Drug Metab Rev 46: 207-223. doi: 10.3109/03602532.2014.882354
|
| [11] | Hane F, Drolle E, Gaikwad R, et al. (2011) Amyloid-beta aggregation on model lipid membranes: an atomic force microscopy study. J Alzheimers Dis 26: 485-494. |
| [12] |
Lal R, Lin H, Quist AP (2007) Amyloid beta ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim Biophys Acta 1768: 1966-1975. doi: 10.1016/j.bbamem.2007.04.021
|
| [13] |
Arispe N, Pollard HB, Rojas E (1993) Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membranes. Proc Natl Acad Sci U S A 90: 10573-10577. doi: 10.1073/pnas.90.22.10573
|
| [14] |
Demuro A, Smith M, Parker I (2011) Single-channel Ca(2+) imaging implicates Abeta1-42 amyloid pores in Alzheimer's disease pathology. J Cell Biol 195: 515-524. doi: 10.1083/jcb.201104133
|
| [15] | Plant LD, Boyle JP, Smith IF, et al. (2003) The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J Neurosci 23: 5531-5535. |
| [16] |
Giuffrida ML, Caraci F, Pignataro B, et al. (2009) Beta-amyloid monomers are neuroprotective. J Neurosci 29: 10582-10587. doi: 10.1523/JNEUROSCI.1736-09.2009
|
| [17] |
Hardy J, Selkoe DJ (2002) The Amyloid Hypothesis of Alzheimer's Disease: Progress and Problems on the Road to Therapeutics. Science 297: 353-356. doi: 10.1126/science.1072994
|
| [18] |
Brambilla D, Verpillot R, Le Droumaguet B, et al. (2012) PEGylated nanoparticles bind to and alter amyloid-beta peptide conformation: toward engineering of functional nanomedicines for Alzheimer's disease. ACS Nano 6: 5897-5908. doi: 10.1021/nn300489k
|
| [19] |
Gobbi M, Re F, Canovi M, et al. (2010) Lipid-based nanoparticles with high binding affinity for amyloid-beta1-42 peptide. Biomaterials 31: 6519-6529. doi: 10.1016/j.biomaterials.2010.04.044
|
| [20] |
Cheng KK, Yeung CF, Ho SW, et al. (2013) Highly stabilized curcumin nanoparticles tested in an in vitro blood-brain barrier model and in Alzheimer's disease Tg2576 mice. Aaps j 15: 324-336. doi: 10.1208/s12248-012-9444-4
|
| [21] |
Cheng X, van Breemen RB (2005) Mass Spectrometry-Based Screening for Inhibitors of β-Amyloid Protein Aggregation. Anal Chem 77: 7012-7015. doi: 10.1021/ac050556a
|
| [22] |
He H, Dong W, Huang F (2010) Anti-Amyloidogenic and Anti-Apoptotic Role of Melatonin in Alzheimer Disease. Curr Neuropharmacol 8: 211-217. doi: 10.2174/157015910792246137
|
| [23] |
Lin L, Huang Q-X, Yang S-S, et al. (2013) Melatonin in Alzheimer's Disease. Int J Mol Sci 14: 14575-14593. doi: 10.3390/ijms140714575
|
| [24] | Rosales-Corral S, Acuna-Castroviejo D, Tan DX, et al. (2012) Accumulation of Exogenous Amyloid-Beta Peptide in Hippocampal Mitochondria Causes Their Dysfunction: A Protective Role for Melatonin. Oxid Med Cell Longev 2012: 843649. |
| [25] |
Adolfsson O, Pihlgren M, Toni N, et al. (2012) An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci 32: 9677-9689. doi: 10.1523/JNEUROSCI.4742-11.2012
|
| [26] |
DeMattos RB, Bales KR, Cummins DJ, et al. (2001) Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 98: 8850-8855. doi: 10.1073/pnas.151261398
|
| [27] |
Legleiter J, Czilli DL, Gitter B, et al. (2004) Effect of different anti-Abeta antibodies on Abeta fibrillogenesis as assessed by atomic force microscopy. J Mol Biol 335: 997-1006. doi: 10.1016/j.jmb.2003.11.019
|
| [28] |
Lemere CA (2013) Immunotherapy for Alzheimer's disease: hoops and hurdles. Mol Neurodegener 8: 36. doi: 10.1186/1750-1326-8-36
|
| [29] |
Robert R, Wark KL (2012) Engineered antibody approaches for Alzheimer's disease immunotherapy. Arch Biochem Biophys 526: 132-138. doi: 10.1016/j.abb.2012.02.022
|
| [30] |
Wilcock DM, Rojiani A, Rosenthal A, et al. (2004) Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation 1: 24. doi: 10.1186/1742-2094-1-24
|
| [31] |
Findeis MA, Musso GM, Arico-Muendel CC, et al. (1999) Modified-peptide inhibitors of amyloid beta-peptide polymerization. Biochemistry 38: 6791-6800. doi: 10.1021/bi982824n
|
| [32] |
Ghanta J, Shen CL, Kiessling LL, et al. (1996) A strategy for designing inhibitors of beta-amyloid toxicity. J Biol Chem 271: 29525-29528. doi: 10.1074/jbc.271.47.29525
|
| [33] |
Hane FT, Lee BY, Petoyan A, et al. (2014) Testing synthetic amyloid-beta aggregation inhibitor using single molecule atomic force spectroscopy. Biosens Bioelectron 54: 492-498. doi: 10.1016/j.bios.2013.10.060
|
| [34] |
Pallitto MM, Ghanta J, Heinzelman P, et al. (1999) Recognition sequence design for peptidyl modulators of beta-amyloid aggregation and toxicity. Biochemistry 38: 3570-3578. doi: 10.1021/bi982119e
|
| [35] |
Tjernberg LO, Naslund J, Lindqvist F, et al. (1996) Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J Biol Chem 271: 8545-8548. doi: 10.1074/jbc.271.15.8545
|
| [36] |
Morgan D, Diamond DM, Gottschall PE, et al. (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408: 982-985. doi: 10.1038/35050116
|
| [37] |
Orgogozo JM, Gilman S, Dartigues JF, et al. (2003) Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61: 46-54. doi: 10.1212/01.WNL.0000073623.84147.A8
|
| [38] |
Wildsmith KR, Holley M, Savage JC, et al. (2013) Evidence for impaired amyloid beta clearance in Alzheimer's disease. Alzheimers Res Ther 5: 33. doi: 10.1186/alzrt187
|
| [39] |
Evin G, Hince C (2013) BACE1 as a therapeutic target in Alzheimer's disease: rationale and current status. Drugs Aging 30: 755-764. doi: 10.1007/s40266-013-0099-3
|
| [40] | Wolfe MS (2012) gamma-Secretase inhibitors and modulators for Alzheimer's disease. J Neurochem 120 Suppl 1: 89-98. |
| [41] | Atwal JK, Chen Y, Chiu C, et al. (2011) A therapeutic antibody targeting BACE1 inhibits amyloid-beta production in vivo. Sci Transl Med 3: 84ra43. |
| [42] |
Kao SC, Krichevsky AM, Kosik KS, et al. (2004) BACE1 suppression by RNA interference in primary cortical neurons. J Biol Chem 279: 1942-1949. doi: 10.1074/jbc.M309219200
|
| [43] |
Singer O, Marr RA, Rockenstein E, et al. (2005) Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci 8: 1343-1349. doi: 10.1038/nn1531
|
| [44] |
Vassar R (2014) BACE1 inhibitor drugs in clinical trials for Alzheimer's disease. Alzheimers Res Ther 6: 89. doi: 10.1186/s13195-014-0089-7
|
| [45] |
Luo Y, Bolon B, Kahn S, et al. (2001) Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci 4: 231-232. doi: 10.1038/85059
|
| [46] |
Jacobsen H, Ozmen L, Caruso A, et al. (2014) Combined treatment with a BACE inhibitor and anti-Abeta antibody gantenerumab enhances amyloid reduction in APPLondon mice. J Neurosci 34: 11621-11630. doi: 10.1523/JNEUROSCI.1405-14.2014
|
| [47] |
Morgan D (2005) Mechanisms of A beta plaque clearance following passive A beta immunization. Neurodegener Dis 2: 261-266. doi: 10.1159/000090366
|
| [48] |
Banks WA, Terrell B, Farr SA, et al. (2002) Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer's disease. Peptides 23: 2223-2226. doi: 10.1016/S0196-9781(02)00261-9
|
| [49] |
Banks WA (2012) Drug delivery to the brain in Alzheimer's disease: consideration of the blood-brain barrier. Adv Drug Deliv Rev 64: 629-639. doi: 10.1016/j.addr.2011.12.005
|
| [50] |
Pfeifer LA, White LR, Ross GW, et al. (2002) Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58: 1629-1634. doi: 10.1212/WNL.58.11.1629
|
| [51] |
Racke MM, Boone LI, Hepburn DL, et al. (2005) Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci 25: 629-636. doi: 10.1523/JNEUROSCI.4337-04.2005
|
| [52] | Wilcock DM, Colton CA (2008) Anti-amyloid-beta immunotherapy in Alzheimer's disease: relevance of transgenic mouse studies to clinical trials. J Alzheimers Dis 15: 555-569. |
| [53] |
Doody RS, Thomas RG, Farlow M, et al. (2014) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med 370: 311-321. doi: 10.1056/NEJMoa1312889
|
| [54] |
Panza F, Solfrizzi V, Imbimbo BP, et al. (2014) Amyloid-based immunotherapy for Alzheimer's disease in the time of prevention trials: the way forward. Expert Rev Clin Immunol 10: 405-419. doi: 10.1586/1744666X.2014.883921
|
| [55] |
Salloway S, Sperling R, Fox NC, et al. (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl J Med 370: 322-333. doi: 10.1056/NEJMoa1304839
|
| [56] |
Tayeb HO, Murray ED, Price BH, et al. (2013) Bapineuzumab and solanezumab for Alzheimer's disease: is the 'amyloid cascade hypothesis' still alive? Expert Opin Biol Ther 13: 1075-1084. doi: 10.1517/14712598.2013.789856
|
| [57] |
Garber K (2012) Genentech's Alzheimer's antibody trial to study disease prevention. Nat Biotechnol 30: 731-732. doi: 10.1038/nbt0812-731
|
| [58] |
Panza F, Solfrizzi V, Imbimbo BP, et al. (2014) Efficacy and safety studies of gantenerumab in patients with Alzheimer's disease. Expert Rev Neurother 14: 973-986. doi: 10.1586/14737175.2014.945522
|
| [59] |
Kumar J, Sim V (2014) D-amino acid-based peptide inhibitors as early or preventative therapy in Alzheimer disease. Prion 8: 119-124. doi: 10.4161/pri.28220
|
| [60] |
Porat Y, Mazor Y, Efrat S, et al. (2004) Inhibition of islet amyloid polypeptide fibril formation: a potential role for heteroaromatic interactions. Biochemistry 43: 14454-14462. doi: 10.1021/bi048582a
|
| [61] |
Austen BM, Paleologou KE, Ali SA, et al. (2008) Designing peptide inhibitors for oligomerization and toxicity of Alzheimer's beta-amyloid peptide. Biochemistry 47: 1984-1992. doi: 10.1021/bi701415b
|
| [62] |
Taylor M, Moore S, Mayes J, et al. (2010) Development of a proteolytically stable retro-inverso peptide inhibitor of beta-amyloid oligomerization as a potential novel treatment for Alzheimer's disease. Biochemistry 49: 3261-3272. doi: 10.1021/bi100144m
|
| [63] | Roy S (2010) Designing Novel Peptidic Inhibitors of Beta Amyloid Oligomerization Calgary, Alberta: University of Calgary. 164 p. |
| [64] | Gordon DJ, Tappe R, Meredith SC (2002) Design and characterization of a membrane permeable N-methyl amino acid-containing peptide that inhibits Abeta1-40 fibrillogenesis. J Pept Res 60: 37-55. |
| [65] |
Parthsarathy V, McClean PL, Holscher C, et al. (2013) A novel retro-inverso peptide inhibitor reduces amyloid deposition, oxidation and inflammation and stimulates neurogenesis in the APPswe/PS1DeltaE9 mouse model of Alzheimer's disease. PLoS One 8: e54769. doi: 10.1371/journal.pone.0054769
|
| [66] | Yu YJ, Zhang Y, Kenrick M, et al. (2011) Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med 3: 84ra44. |
| [67] |
Xiong N, Dong X-Y, Zheng J, et al. (2015) Design of LVFFARK and LVFFARK-functionalized nanoparticles for inhibiting amyloid β-protein fibrillation and cytotoxicity. ACS Appl Mater Interfaces 7: 5650-5662. doi: 10.1021/acsami.5b00915
|
| [68] |
Taylor M, Moore S, Mourtas S, et al. (2011) Effect of curcumin-associated and lipid ligand-functionalized nanoliposomes on aggregation of the Alzheimer's Abeta peptide. Nanomedicine 7: 541-550. doi: 10.1016/j.nano.2011.06.015
|
| [69] | Armstrong RA (2014) A critical analysis of the 'amyloid cascade hypothesis'. Folia Neuropathol 52: 211-225. |
| [70] | Castello MA, Soriano S (2014) On the origin of Alzheimer's disease. Trials and tribulations of the amyloid hypothesis. Ageing Res Rev 13: 10-12. |
| [71] |
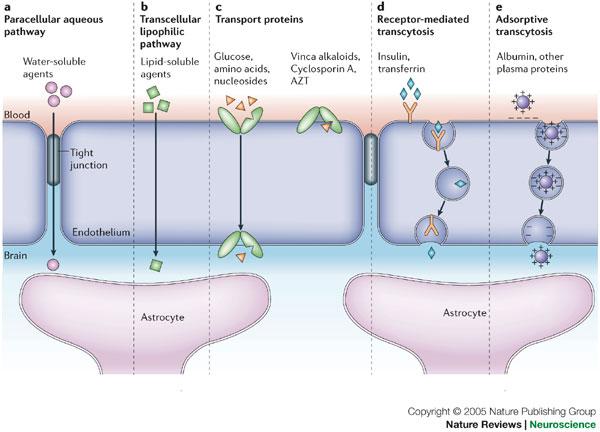
Chen Y, Liu L (2012) Modern methods for delivery of drugs across the blood-brain barrier. Adv Drug Deliv Rev 64: 640-665. doi: 10.1016/j.addr.2011.11.010
|
| [72] |
Pardridge WM (2003) Blood-brain barrier drug targeting: the future of brain drug development. Mol Interv 3: 90-105, 151. doi: 10.1124/mi.3.2.90
|
| [73] |
Abbott NJ, Patabendige AA, Dolman DE, et al. (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37: 13-25. doi: 10.1016/j.nbd.2009.07.030
|
| [74] |
Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7: 41-53. doi: 10.1038/nrn1824
|
| [75] |
Mehta DC, Short JL, Nicolazzo JA (2013) Memantine transport across the mouse blood-brain barrier is mediated by a cationic influx H+ antiporter. Mol Pharm 10: 4491-4498. doi: 10.1021/mp400316e
|
| [76] |
Kim MH, Maeng HJ, Yu KH, et al. (2010) Evidence of carrier-mediated transport in the penetration of donepezil into the rat brain. J Pharm Sci 99: 1548-1566. doi: 10.1002/jps.21895
|
| [77] |
Cirrito JR, Deane R, Fagan AM, et al. (2005) P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest 115: 3285-3290. doi: 10.1172/JCI25247
|
| [78] |
Deane R, Wu Z, Zlokovic BV (2004) RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 35: 2628-2631. doi: 10.1161/01.STR.0000143452.85382.d1
|
| [79] | Guillemin GJ, Brew BJ (2004) Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol 75: 388-397. |
| [80] |
Bowman GL, Kaye JA, Moore M, et al. (2007) Blood-brain barrier impairment in Alzheimer disease: stability and functional significance. Neurology 68: 1809-1814. doi: 10.1212/01.wnl.0000262031.18018.1a
|
| [81] | Claudio L (1996) Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer's disease patients. Acta Neuropathol 91: 6-14. |
| [82] | Kalaria RN (1992) The blood-brain barrier and cerebral microcirculation in Alzheimer disease. Cerebrovasc Brain Metab Rev 4: 226-260. |
| [83] |
Marco S, Skaper SD (2006) Amyloid beta-peptide1-42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci Lett 401: 219-224. doi: 10.1016/j.neulet.2006.03.047
|
| [84] |
Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci 28: 202-208. doi: 10.1016/j.tins.2005.02.001
|
| [85] |
Wisniewski HM, Vorbrodt AW, Wegiel J (1997) Amyloid angiopathy and blood-brain barrier changes in Alzheimer's disease. Ann N Y Acad Sci 826: 161-172. doi: 10.1111/j.1749-6632.1997.tb48468.x
|
| [86] |
Kalaria RN, Sromek SM, Grahovac I, et al. (1992) Transferrin receptors of rat and human brain and cerebral microvessels and their status in Alzheimer's disease. Brain Res 585: 87-93. doi: 10.1016/0006-8993(92)91193-I
|
| [87] |
Jones AR, Shusta EV (2007) Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm Res 24: 1759-1771. doi: 10.1007/s11095-007-9379-0
|
| [88] |
Gatter KC, Brown G, Trowbridge IS, et al. (1983) Transferrin receptors in human tissues: their distribution and possible clinical relevance. J Clin Pathol 36: 539-545. doi: 10.1136/jcp.36.5.539
|
| [89] | Lee HJ, Engelhardt B, Lesley J, et al. (2000) Targeting rat anti-mouse transferrin receptor monoclonal antibodies through blood-brain barrier in mouse. J Pharmacol Exp Ther 292: 1048-1052. |
| [90] |
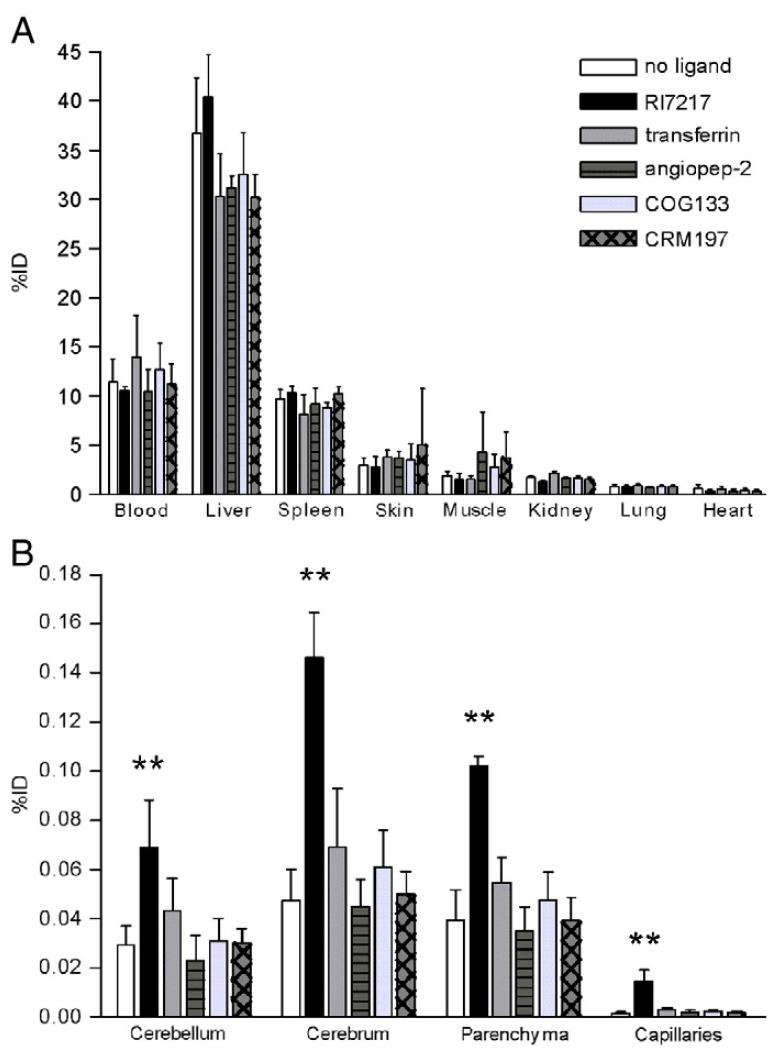
Prades R, Guerrero S, Araya E, et al. (2012) Delivery of gold nanoparticles to the brain by conjugation with a peptide that recognizes the transferrin receptor. Biomaterials 33: 7194-7205. doi: 10.1016/j.biomaterials.2012.06.063
|
| [91] | Zhao WQ, De Felice FG, Fernandez S, et al. (2008) Amyloid beta oligomers induce impairment of neuronal insulin receptors. Faseb J 22: 246-260. |
| [92] | Xie L, Helmerhorst E, Taddei K, et al. (2002) Alzheimer's beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci 22: Rc221. |
| [93] |
Markoutsa E, Papadia K, Giannou AD, et al. (2014) Mono and dually decorated nanoliposomes for brain targeting, in vitro and in vivo studies. Pharm Res 31: 1275-1289. doi: 10.1007/s11095-013-1249-3
|
| [94] | Re F, Cambianica I, Sesana S, et al. (2010) Functionalization with ApoE-derived peptides enhances the interaction with brain capillary endothelial cells of nanoliposomes binding amyloid-beta peptide. J Biotechnol 156: 341-346. |
| [95] |
Gaillard PJ, Visser CC, de Boer AG (2005) Targeted delivery across the blood-brain barrier. Expert Opin Drug Deliv 2: 299-309. doi: 10.1517/17425247.2.2.299
|
| [96] |
Lentz TL (1990) Rabies virus binding to an acetylcholine receptor alpha-subunit peptide. J Mol Recognit 3: 82-88. doi: 10.1002/jmr.300030205
|
| [97] |
Liu Y, Huang R, Han L, et al. (2009) Brain-targeting gene delivery and cellular internalization mechanisms for modified rabies virus glycoprotein RVG29 nanoparticles. Biomaterials 30: 4195-4202. doi: 10.1016/j.biomaterials.2009.02.051
|
| [98] | Couch JA, Yu YJ, Zhang Y, et al. (2013) Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med 5: 183ra157, 181-112. |
| [99] |
Bien-Ly N, Yu YJ, Bumbaca D, et al. (2014) Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J Exp Med 211: 233-244. doi: 10.1084/jem.20131660
|
| [100] | Yu YJ, Atwal JK, Zhang Y, et al. (2014) Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med 6: 261ra154. |
| [101] |
Niewoehner J, Bohrmann B, Collin L, et al. (2014) Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 81: 49-60. doi: 10.1016/j.neuron.2013.10.061
|
| [102] |
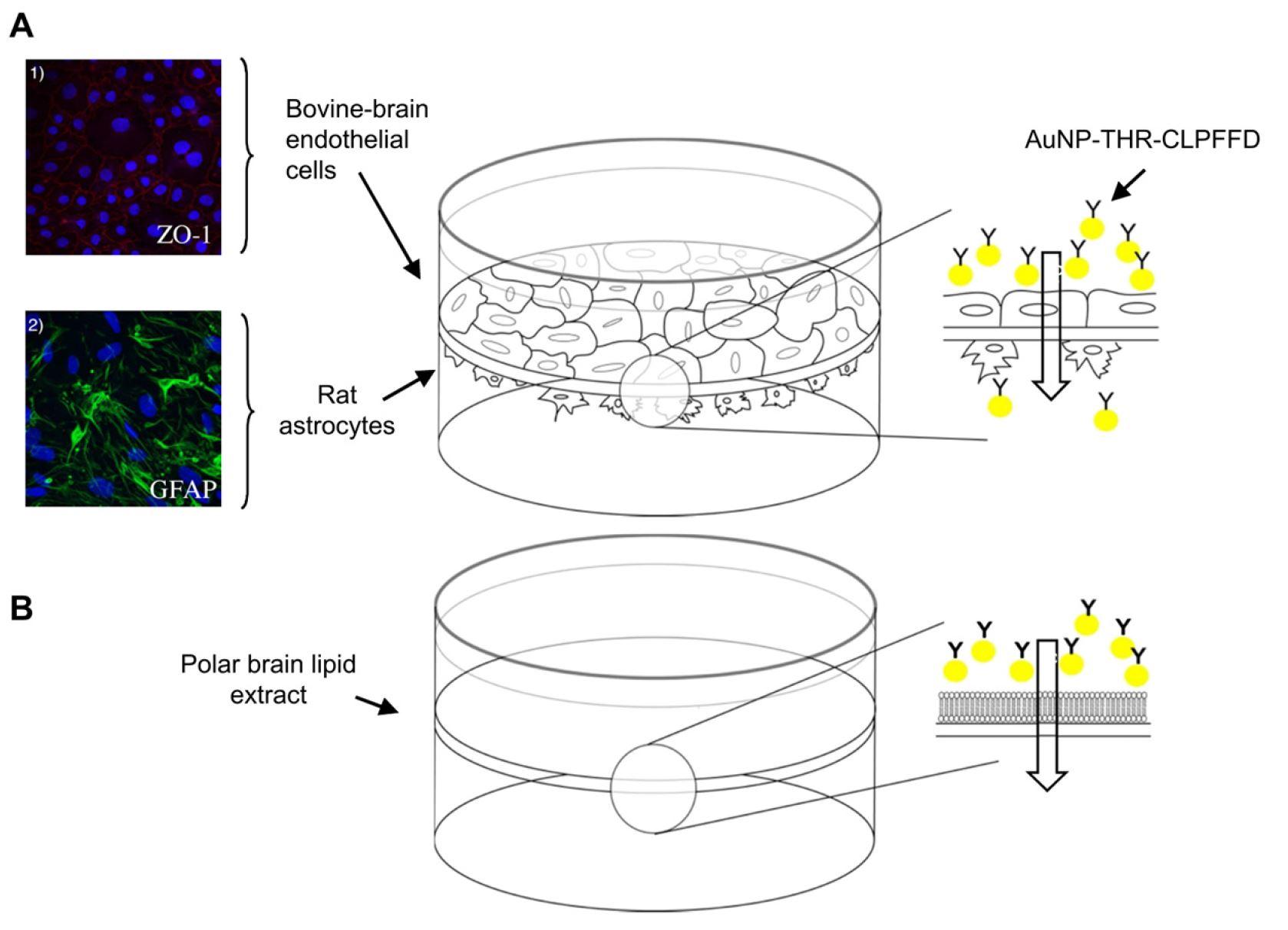
van Rooy I, Mastrobattista E, Storm G, et al. (2011) Comparison of five different targeting ligands to enhance accumulation of liposomes into the brain. J Control Release 150: 30-36. doi: 10.1016/j.jconrel.2010.11.014
|
| [103] |
Zhang C, Wan X, Zheng X, et al. (2014) Dual-functional nanoparticles targeting amyloid plaques in the brains of Alzheimer's disease mice. Biomaterials 35: 456-465. doi: 10.1016/j.biomaterials.2013.09.063
|
| [104] |
Zhang C, Zheng X, Wan X, et al. (2014) The potential use of H102 peptide-loaded dual-functional nanoparticles in the treatment of Alzheimer's disease. J Control Release 192: 317-324. doi: 10.1016/j.jconrel.2014.07.050
|
| [105] | Salvati E, Re F, Sesana S, et al. (2013) Liposomes functionalized to overcome the blood-brain barrier and to target amyloid-beta peptide: the chemical design affects the permeability across an in vitro model. Int J Nanomedicine 8: 1749-1758. |
| [106] |
Markoutsa E, Papadia K, Clemente C, et al. (2012) Anti-Abeta-MAb and dually decorated nanoliposomes: effect of Abeta1-42 peptides on interaction with hCMEC/D3 cells. Eur J Pharm Biopharm 81: 49-56. doi: 10.1016/j.ejpb.2012.02.006
|
| [107] |
Herve F, Ghinea N, Scherrmann JM (2008) CNS delivery via adsorptive transcytosis. AAPS J 10: 455-472. doi: 10.1208/s12248-008-9055-2
|
| [108] | Pardridge WM, Buciak JL, Kang YS, et al. (1993) Protamine-mediated transport of albumin into brain and other organs of the rat. Binding and endocytosis of protamine-albumin complex by microvascular endothelium. J Clin Invest 92: 2224-2229. |
| [109] |
Bickel U (1995) Antibody delivery through the blood-brain barrier. Adv Drug Deliv Rev 15: 53-72. doi: 10.1016/0169-409X(95)00005-R
|
| [110] |
Bechara C, Sagan S (2013) Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett 587: 1693-1702. doi: 10.1016/j.febslet.2013.04.031
|
| [111] |
Jaruszewski KM, Ramakrishnan S, Poduslo JF, et al. (2012) Chitosan enhances the stability and targeting of immuno-nanovehicles to cerebro-vascular deposits of Alzheimer's disease amyloid protein. Nanomedicine 8: 250-260. doi: 10.1016/j.nano.2011.06.008
|
| [112] |
Parhamifar L, Sime W, Yudina Y, et al. (2010) Ligand-induced tyrosine phosphorylation of cysteinyl leukotriene receptor 1 triggers internalization and signaling in intestinal epithelial cells. PLoS One 5: e14439. doi: 10.1371/journal.pone.0014439
|
| [113] |
Malhotra M, Tomaro-Duchesneau C, Prakash S (2013) Synthesis of TAT peptide-tagged PEGylated chitosan nanoparticles for siRNA delivery targeting neurodegenerative diseases. Biomaterials 34: 1270-1280. doi: 10.1016/j.biomaterials.2012.10.013
|
| [114] |
Sarvaiya J, Agrawal YK (2015) Chitosan as a suitable nanocarrier material for anti-Alzheimer drug delivery. Int J Biol Macromol 72: 454-465. doi: 10.1016/j.ijbiomac.2014.08.052
|
| [115] |
Gao Y, Wang ZY, Zhang J, et al. (2014) RVG-peptide-linked trimethylated chitosan for delivery of siRNA to the brain. Biomacromolecules 15: 1010-1018. doi: 10.1021/bm401906p
|
| [116] |
Sharma G, Modgil A, Layek B, et al. (2013) Cell penetrating peptide tethered bi-ligand liposomes for delivery to brain in vivo: Biodistribution and transfection. J Control Release 167: 1-10. doi: 10.1016/j.jconrel.2013.01.016
|
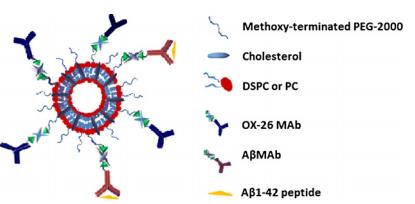
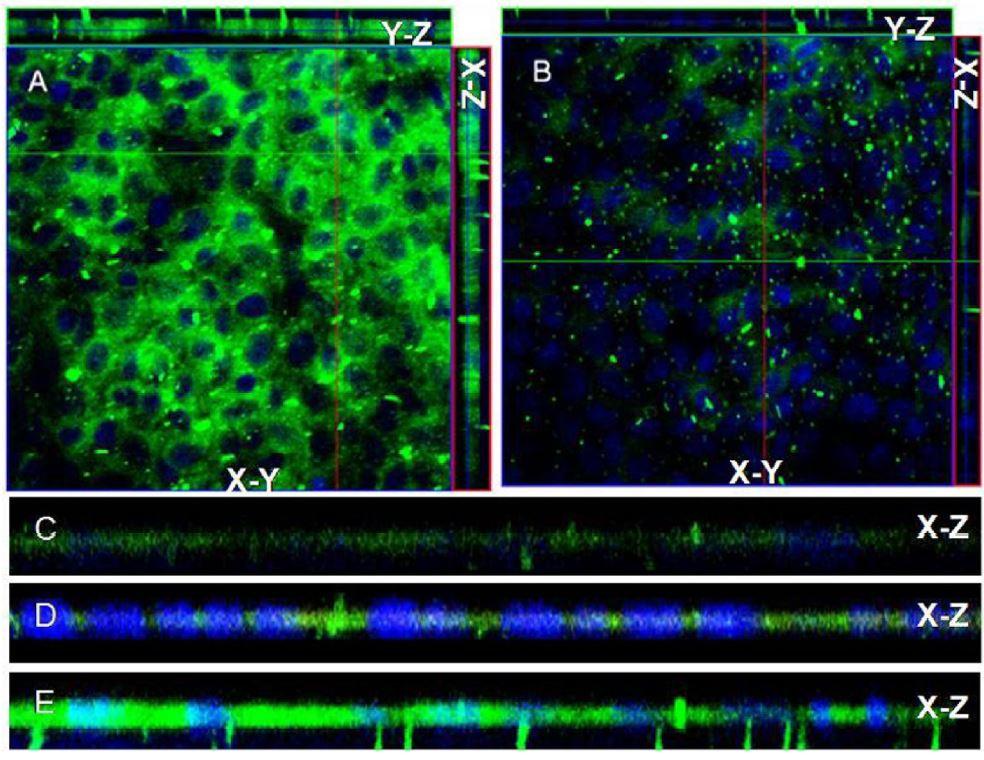
Figures(6)

Morgan Robinson, Brenda Yasie Lee, Zoya Leonenko. Drugs and drug delivery systems targeting amyloid-β in Alzheimer's disease[J]. AIMS Molecular Science, 2015, 2(3): 332-358. doi: 10.3934/molsci.2015.3.332







 DownLoad:
DownLoad: