Citation: Thomas Rauen, Rose Tanui, Christof Grewer. Structural and functional dynamics of Excitatory Amino Acid Transporters (EAAT)[J]. AIMS Molecular Science, 2014, 1(3): 99-125. doi: 10.3934/molsci.2014.3.99

| [1] |
Zerangue N, Kavanaugh MP (1996) Flux coupling in a neuronal glutamate transporter. Nature 383: 634-637. doi: 10.1038/383634a0

|
| [2] |
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65: 1-105. doi: 10.1016/S0301-0082(00)00067-8
|
| [3] | Hertz L (1979) Functional interactions between neurons and astrocytes. I. Turnover and metabolism of putative amino acid transmitters. ProgNeurobiol 13: 277-323. |
| [4] |
Broer S, Brookes N (2001) Transfer of glutamine between astrocytes and neurons. J Neurochem 77: 705-719. doi: 10.1046/j.1471-4159.2001.00322.x
|
| [5] | Drejer J, Larsson OM, Schousboe A (1982) Characterization of L-glutamate uptake into and release from astrocytes and neurons cultured from differnt brain regions. ExpBrain Res 47: 259-269. |
| [6] | Schousboe A, Hertz L (1981) Role of astroglial cells in glutamate homeostasis. Adv Biochem Psychopharmacol 27: 103-113. |
| [7] | Rauen T, Taylor WR, Kuhlbrodt K, et al. (1998) High-affinity glutamate transporters in the rat retina: a major role of the glial glutamate transporter GLAST-1 in transmitter clearance. Cell Tissue Res 291: 19-31. |
| [8] |
Rauen T, Wiessner M (2000) Fine tuning of glutamate uptake and degradation in glial cells: common transcriptional regulation of GLAST1 and GS. Neurochem Int 37: 179-189. doi: 10.1016/S0197-0186(00)00021-8
|
| [9] |
Furness DN, Dehnes Y, Akhtar AQ, et al. (2008) A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience 157: 80-94. doi: 10.1016/j.neuroscience.2008.08.043
|
| [10] |
Pines G, Danbolt NC, Bjoras M, et al. (1992) Cloning and expression of a rat brain L-glutamate transporter. Nature 360: 464-467. doi: 10.1038/360464a0
|
| [11] |
Storck T, Schulte S, Hofmann K, et al. (1992) Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci U S A 89: 10955-10959. doi: 10.1073/pnas.89.22.10955
|
| [12] | Arriza JL, Fairman WA, Wadiche JI, et al. (1994) Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14: 5559-5569. |
| [13] |
Tanaka K, Watase K, Manabe T, et al. (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276: 1699-1702. doi: 10.1126/science.276.5319.1699
|
| [14] |
Bjornsen LP, Hadera MG, Zhou Y, et al. (2014) The GLT-1 (EAAT2; slc1a2) glutamate transporter is essential for glutamate homeostasis in the neocortex of the mouse. J Neurochem 128: 641-649. doi: 10.1111/jnc.12509
|
| [15] |
Rauen T, Wiessner M, Sullivan R, et al. (2004) A new GLT1 splice variant: cloning and immunolocalization of GLT1c in the mammalian retina and brain. Neurochem Int 45: 1095-1106. doi: 10.1016/j.neuint.2004.04.006
|
| [16] |
Sullivan R, Rauen T, Fischer F, et al. (2004) Cloning, transport properties, and differential localization of two splice variants of GLT-1 in the rat CNS: Implications for CNS glutamate homeostasis. Glia 45: 155-169. doi: 10.1002/glia.10317
|
| [17] |
Lee A, Anderson AR, Beasley SJ, et al. (2012) A new splice variant of the glutamate-aspartate transporter: cloning and immunolocalization of GLAST1c in rat, pig and human brains. J Chem Neuroanat 43: 52-63. doi: 10.1016/j.jchemneu.2011.10.005
|
| [18] |
Grewer C, Gameiro A, Rauen T (2014) SLC1 glutamate transporters. Pflugers Arch 466: 3-24. doi: 10.1007/s00424-013-1397-7
|
| [19] |
Rauen T (2000) Diversity of glutamate transporter expression and function in the mammalian retina. Amino Acids 19: 53-62. doi: 10.1007/s007260070033
|
| [20] |
Rauen T, Kanner BI (1994) Localization of the glutamate transporter GLT-1 in rat and macaque monkey retinae. Neurosci Lett 169: 137-140. doi: 10.1016/0304-3940(94)90375-1
|
| [21] |
Wiessner M, Fletcher EL, Fischer F, et al. (2002) Localization and possible function of the glutamate transporter, EAAC1, in the rat retina. Cell Tissue Res 310: 31-40. doi: 10.1007/s00441-002-0612-1
|
| [22] |
Holmseth S, Dehnes Y, Huang YH, et al. (2012) The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J Neurosci 32: 6000-6013. doi: 10.1523/JNEUROSCI.5347-11.2012
|
| [23] | Dehnes Y, Chaudhry FA, Ullensvang K, et al. (1998) The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci 18: 3606-3619. |
| [24] |
Mim C, Balani P, Rauen T, et al. (2005) The Glutamate Transporter Subtypes EAAT4 and EAATs 1-3 Transport Glutamate with Dramatically Different Kinetics and Voltage Dependence but Share a Common Uptake Mechanism. J Gen Physiol 126: 571-589. doi: 10.1085/jgp.200509365
|
| [25] |
Gincel D, Regan MR, Jin L, et al. (2007) Analysis of cerebellar Purkinje cells using EAAT4 glutamate transporter promoter reporter in mice generated via bacterial artificial chromosome-mediated transgenesis. Exp Neurol 203: 205-212. doi: 10.1016/j.expneurol.2006.08.016
|
| [26] |
Kovermann P, Machtens JP, Ewers D, et al. (2010) A conserved aspartate determines pore properties of anion channels associated with excitatory amino acid transporter 4 (EAAT4). J Biol Chem 285: 23676-23686. doi: 10.1074/jbc.M110.126557
|
| [27] |
Arriza JL, Eliasof S, Kavanaugh MP, et al. (1997) Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci U S A 94: 4155-4160. doi: 10.1073/pnas.94.8.4155
|
| [28] |
Wersinger E, Schwab Y, Sahel JA, et al. (2006) The glutamate transporter EAAT5 works as a presynaptic receptor in mouse rod bipolar cells. J Physiol 577: 221-234. doi: 10.1113/jphysiol.2006.118281
|
| [29] |
Gameiro A, Braams S, Rauen T, et al. (2011) The Discovery of Slowness: Low-Capacity Transport and Slow Anion Channel Gating by the Glutamate Transporter EAAT5. Biophysical journal 100: 2623-2632. doi: 10.1016/j.bpj.2011.04.034
|
| [30] | Hediger MA, Kanai Y, You G, et al. (1995) Mammalian ion-coupled solute transporters. JPhysiolLond 482: 7S-17S. |
| [31] |
Bailey CG, Ryan RM, Thoeng AD, et al. (2011) Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria. J Clin Invest 121: 446-453. doi: 10.1172/JCI44474
|
| [32] |
Duerson K, Woltjer RL, Mookherjee P, et al. (2009) Detergent-insoluble EAAC1/EAAT3 aberrantly accumulates in hippocampal neurons of Alzheimer's disease patients. Brain Pathol 19: 267-278. doi: 10.1111/j.1750-3639.2008.00186.x
|
| [33] |
Revett TJ, Baker GB, Jhamandas J, et al. (2013) Glutamate system, amyloid ss peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci 38: 6-23. doi: 10.1503/jpn.110190
|
| [34] | Rothstein JD (2009) Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol 65 Suppl 1: S3-9. |
| [35] |
Lang UE, Borgwardt S (2013) Molecular Mechanisms of Depression: Perspectives on New Treatment Strategies. Cell Physiol Biochem 31: 761-777. doi: 10.1159/000350094
|
| [36] |
Crino PB, Jin H, Shumate MD, et al. (2002) Increased expression of the neuronal glutamate transporter (EAAT3/EAAC1) in hippocampal and neocortical epilepsy. Epilepsia 43: 211-218. doi: 10.1046/j.1528-1157.2002.35001.x
|
| [37] |
Estrada-Sanchez AM, Rebec GV (2012) Corticostriatal dysfunction and glutamate transporter 1 (GLT1) in Huntington's disease: interactions between neurons and astrocytes. Basal Ganglia 2: 57-66. doi: 10.1016/j.baga.2012.04.029
|
| [38] | Rao VL, Dogan A, Todd KG, et al. (2001) Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J Neurosci 21: 1876-1883. |
| [39] |
Grewer C, Gameiro A, Zhang Z, et al. (2008) Glutamate forward and reverse transport: from molecular mechanism to transporter-mediated release after ischemia. IUBMB Life 60: 609-619. doi: 10.1002/iub.98
|
| [40] |
Ketheeswaranathan P, Turner NA, Spary EJ, et al. (2011) Changes in glutamate transporter expression in mouse forebrain areas following focal ischemia. Brain Res 1418: 93-103. doi: 10.1016/j.brainres.2011.08.029
|
| [41] |
Seki Y, Feustel PJ, Keller RW, et al. (1999) Inhibition of ischemia-induced glutamate release in rat striatum by dihydrokinate and an anion channel blocker. Stroke 30: 433-440. doi: 10.1161/01.STR.30.2.433
|
| [42] | Azami Tameh A, Clarner T, Beyer C, et al. (2013) Regional regulation of glutamate signaling during cuprizone-induced demyelination in the brain. Ann Anat. |
| [43] |
Karlsson RM, Tanaka K, Heilig M, et al. (2008) Loss of glial glutamate and aspartate transporter (excitatory amino acid transporter 1) causes locomotor hyperactivity and exaggerated responses to psychotomimetics: rescue by haloperidol and metabotropic glutamate 2/3 agonist. Biol Psychiatry 64: 810-814. doi: 10.1016/j.biopsych.2008.05.001
|
| [44] |
Karlsson RM, Tanaka K, Saksida LM, et al. (2009) Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology 34: 1578-1589. doi: 10.1038/npp.2008.215
|
| [45] |
Adamczyk A, Gause CD, Sattler R, et al. (2011) Genetic and functional studies of a missense variant in a glutamate transporter, SLC1A3, in Tourette syndrome. Psychiatr Genet 21: 90-97. doi: 10.1097/YPG.0b013e328341a307
|
| [46] |
Reyes N, Ginter C, Boudker O (2009) Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462: 880-885. doi: 10.1038/nature08616
|
| [47] |
Verdon G, Boudker O (2012) Crystal structure of an asymmetric trimer of a bacterial glutamate transporter homolog. Nat Struct Mol Biol 19: 355-357. doi: 10.1038/nsmb.2233
|
| [48] |
Yernool D, Boudker O, Jin Y, et al. (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431: 811-818. doi: 10.1038/nature03018
|
| [49] |
Jardetzky O (1966) Simple allosteric model for membrane pumps. Nature 211: 969-970. doi: 10.1038/211969a0
|
| [50] |
Owe SG, Marcaggi P, Attwell D (2006) The ionic stoichiometry of the GLAST glutamate transporter in salamander retinal glia. J Physiol 577: 591-599. doi: 10.1113/jphysiol.2006.116830
|
| [51] |
Kanai Y, Nussberger S, Romero MF, et al. (1995) Electrogenic properties of the epithelial and neuronal high affinity glutamate transporter. J Biol Chem 270: 16561-16568. doi: 10.1074/jbc.270.28.16561
|
| [52] | Wadiche JI, Kavanaugh MP (1998) Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J Neurosci 18: 7650-7661. |
| [53] | Otis TS, Kavanaugh MP (2000) Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J Neurosci 20: 2749-2757. |
| [54] | Otis TS, Jahr CE (1998) Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J Neurosci 18: 7099-7110. |
| [55] | Bergles DE, Tzingounis AV, Jahr CE (2002) Comparison of coupled and uncoupled currents during glutamate uptake by GLT-1 transporters. J Neurosci 22: 10153-10162. |
| [56] |
Grewer C, Watzke N, Wiessner M, et al. (2000) Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc Natl Acad Sci U S A 97: 9706-9711. doi: 10.1073/pnas.160170397
|
| [57] |
Watzke N, Bamberg E, Grewer C (2001) Early intermediates in the transport cycle of the neuronal excitatory amino acid carrier EAAC1. J Gen Physiol 117: 547-562. doi: 10.1085/jgp.117.6.547
|
| [58] | Mwaura J, Tao Z, James H, et al. (2012) Protonation state of a conserved acidic amino acid involved in Na(+) binding to the glutamate transporter EAAC1. ACS Chem Neurosci 12: 1073-1083. |
| [59] | Diamond JS, Jahr CE (1997) Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci 17: 4672-4687. |
| [60] |
Mim C, Tao Z, Grewer C (2007) Two conformational changes are associated with glutamate translocation by the glutamate transporter EAAC1. Biochemistry 46: 9007-9018. doi: 10.1021/bi7005465
|
| [61] |
Wadiche JI, Arriza JL, Amara SG, et al. (1995) Kinetics of a human glutamate transporter. Neuron 14: 1019-1027. doi: 10.1016/0896-6273(95)90340-2
|
| [62] |
Loo DD, Hazama A, Supplisson S, et al. (1993) Relaxation kinetics of the Na+/glucose cotransporter. Proc Natl Acad Sci U S A 90: 5767-5771. doi: 10.1073/pnas.90.12.5767
|
| [63] | Lu CC, Hilgemann DW (1999) GAT1 (GABA:Na+:Cl-) cotransport function. Kinetic studies in giant Xenopus oocyte membrane patches. J Gen Physiol 114: 445-457. |
| [64] |
Grewer C, Zhang Z, Mwaura J, et al. (2012) Charge compensation mechanism of a Na+-coupled, secondary active glutamate transporter. J Biol Chem 287: 26921-26931. doi: 10.1074/jbc.M112.364059
|
| [65] |
Zhang Z, Tao Z, Gameiro A, et al. (2007) Transport direction determines the kinetics of substrate transport by the glutamate transporter EAAC1. Proc Natl Acad Sci U S A 104: 18025-18030. doi: 10.1073/pnas.0704570104
|
| [66] |
Wadiche JI, Amara SG, Kavanaugh MP (1995) Ion fluxes associated with excitatory amino acid transport. Neuron 15: 721-728. doi: 10.1016/0896-6273(95)90159-0
|
| [67] |
Eliasof S, Jahr CE (1996) Retinal glial cell glutamate transporter is coupled to an anionic conductance. Proc Natl Acad Sci U S A 93: 4153-4158. doi: 10.1073/pnas.93.9.4153
|
| [68] | Billups B, Rossi D, Attwell D (1996) Anion conductance behavior of the glutamate uptake carrier in salamander retinal glial cells. J Neurosci 16: 6722-6731. |
| [69] |
Fairman WA, Vandenberg RJ, Arriza JL, et al. (1995) An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375: 599-603. doi: 10.1038/375599a0
|
| [70] |
Larsson HP, Picaud SA, Werblin FS, et al. (1996) Noise analysis of the glutamate-activated current in photoreceptors. Biophysl J 70: 733-742. doi: 10.1016/S0006-3495(96)79613-3
|
| [71] |
Melzer N, Biela A, Fahlke C (2003) Glutamate modifies ion conduction and voltage-dependent gating of excitatory amino acid transporter-associated anion channels. J Biol Chem 278: 50112-50119. doi: 10.1074/jbc.M307990200
|
| [72] | Picaud SA, Larsson HP, Grant GB, et al. (1995) Glutamate-gated chloride channel with glutamate-transporter-like properties in cone photoreceptors of the tiger salamander. J Neurophys 74: 1760-1771. |
| [73] |
Watzke N, Grewer C (2001) The anion conductance of the glutamate transporter EAAC1 depends on the direction of glutamate transport. FEBS Lett 503: 121-125. doi: 10.1016/S0014-5793(01)02715-6
|
| [74] |
Tao Z, Grewer C (2007) Cooperation of the conserved aspartate 439 and bound amino acid substrate is important for high-affinity Na+ binding to the glutamate transporter EAAC1. J Gen Physiol 129: 331-344. doi: 10.1085/jgp.200609678
|
| [75] |
Boudker O, Ryan RM, Yernool D, et al. (2007) Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445: 387-393. doi: 10.1038/nature05455
|
| [76] |
Cater RJ, Vandenberg RJ, Ryan RM (2014) The domain interface of the human glutamate transporter EAAT1 mediates chloride permeation. Biophys J 107: 621-629. doi: 10.1016/j.bpj.2014.05.046
|
| [77] |
Huang Z, Tajkhorshid E (2008) Dynamics of the extracellular gate and ion-substrate coupling in the glutamate transporter. Biophys J 95: 2292-2300. doi: 10.1529/biophysj.108.133421
|
| [78] |
Shrivastava IH, Jiang J, Amara SG, et al. (2008) Time-resolved mechanism of extracellular gate opening and substrate binding in a glutamate transporter. J Biol Chem 283: 28680-28690. doi: 10.1074/jbc.M800889200
|
| [79] |
Huang Z, Tajkhorshid E (2010) Identification of the third Na+ site and the sequence of extracellular binding events in the glutamate transporter. Biophys J 99: 1416-1425. doi: 10.1016/j.bpj.2010.06.052
|
| [80] |
Bastug T, Heinzelmann G, Kuyucak S, et al. (2012) Position of the third Na+ site in the aspartate transporter GltPh and the human glutamate transporter, EAAT1. PLoS One 7: e33058. doi: 10.1371/journal.pone.0033058
|
| [81] |
Groeneveld M, Slotboom DJ (2010) Na(+):aspartate coupling stoichiometry in the glutamate transporter homologue Glt(Ph). Biochemistry 49: 3511-3513. doi: 10.1021/bi100430s
|
| [82] |
Larsson HP, Wang X, Lev B, et al. (2010) Evidence for a third sodium-binding site in glutamate transporters suggests an ion/substrate coupling model. Proc Natl Acad Sci U S A 107: 13912-13917. doi: 10.1073/pnas.1006289107
|
| [83] |
DeChancie J, Shrivastava IH, Bahar I (2011) The mechanism of substrate release by the aspartate transporter GltPh: insights from simulations. Mol Biosyst 7: 832-842. doi: 10.1039/C0MB00175A
|
| [84] |
Zomot E, Bahar I (2013) Intracellular gating in an inward-facing state of aspartate transporter Glt(Ph) is regulated by the movements of the helical hairpin HP2. J Biol Chem 288: 8231-8237. doi: 10.1074/jbc.M112.438432
|
| [85] |
Heinzelmann G, Kuyucak S (2014) Molecular dynamics simulations of the mammalian glutamate transporter EAAT3. PLoS One 9: e92089. doi: 10.1371/journal.pone.0092089
|
| [86] |
Jiang J, Shrivastava IH, Watts SD, et al. (2011) Large collective motions regulate the functional properties of glutamate transporter trimers. Proc Natl Acad Sci U S A 108: 15141-15146. doi: 10.1073/pnas.1112216108
|
| [87] |
Lezon TR, Bahar I (2012) Constraints imposed by the membrane selectively guide the alternating access dynamics of the glutamate transporter GltPh. Biophys J 102: 1331-1340. doi: 10.1016/j.bpj.2012.02.028
|
| [88] |
Das A, Gur M, Cheng MH, et al. (2014) Exploring the conformational transitions of biomolecular systems using a simple two-state anisotropic network model. PLoS Comput Biol 10: e1003521. doi: 10.1371/journal.pcbi.1003521
|
| [89] |
Stolzenberg S, Khelashvili G, Weinstein H (2012) Structural intermediates in a model of the substrate translocation path of the bacterial glutamate transporter homologue GltPh. J Phys Chem B 116: 5372-5383. doi: 10.1021/jp301726s
|
| [90] |
Grewer C, Watzke N, Rauen T, et al. (2003) Is the glutamate residue Glu-373 the proton acceptor of the excitatory amino acid carrier 1? J Biol Chem 278: 2585-2592. doi: 10.1074/jbc.M207956200
|
| [91] |
Heinzelmann G, Kuyucak S (2014) Molecular Dynamics Simulations Elucidate the Mechanism of Proton Transport in the Glutamate Transporter EAAT3. Biophys J 106: 2675-2683. doi: 10.1016/j.bpj.2014.05.010
|
| [92] |
Grewer C, Jager J, Carpenter BK, et al. (2000) A new photolabile precursor of glycine with improved properties: A tool for chemical kinetic investigations of the glycine receptor. Biochemistry 39: 2063-2070. doi: 10.1021/bi9919652
|
| [93] |
Grewer C, Rauen T (2005) Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J Membr Biol 203: 1-20. doi: 10.1007/s00232-004-0731-6
|
| [94] |
Gegelashvili G, Robinson MB, Trotti D, et al. (2001) Regulation of glutamate transporters in health and disease. Prog Brain Res 132: 267-286. doi: 10.1016/S0079-6123(01)32082-4
|
| [95] |
Santos SD, Carvalho AL, Caldeira MV, et al. (2009) Regulation of AMPA receptors and synaptic plasticity. Neuroscience 158: 105-125. doi: 10.1016/j.neuroscience.2008.02.037
|
| [96] |
Stephenson FA, Cousins SL, Kenny AV (2008) Assembly and forward trafficking of NMDA receptors (Review). Mol Membr Biol 25: 311-320. doi: 10.1080/09687680801971367
|
| [97] | Robinson MB (2002) Regulated trafficking of neurotransmitter transporters: common notes but different melodies. J Neurochem 80: 1-11. |
| [98] |
Gonzalez MI, Robinson MB (2004) Protein KINASE C-Dependent Remodeling of Glutamate Transporter Function. Mol Intervent 4: 48-58. doi: 10.1124/mi.4.1.48
|
| [99] |
Sheldon AL, Robinson MB (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int 51: 333-355. doi: 10.1016/j.neuint.2007.03.012
|
| [100] | Beart PM, O'Shea RD (2007) Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol 150: 5-17. |
| [101] |
Poitry-Yamate CL, Vutskits L, Rauen T (2002) Neuronal-induced and glutamate-dependent activation of glial glutamate transporter function. J Neurochem 82: 987-997. doi: 10.1046/j.1471-4159.2002.01075.x
|
| [102] |
Benediktsson AM, Marrs GS, Tu JC, et al. (2012) Neuronal activity regulates glutamate transporter dynamics in developing astrocytes. Glia 60: 175-188. doi: 10.1002/glia.21249
|
| [103] |
Gonzalez-Gonzalez IM, Garcia-Tardon N, Gimenez C, et al. (2008) PKC-dependent endocytosis of the GLT1 glutamate transporter depends on ubiquitylation of lysines located in a C-terminal cluster. Glia 56: 963-974. doi: 10.1002/glia.20670
|
| [104] |
Sheldon AL, Gonzalez MI, Krizman-Genda EN, et al. (2008) Ubiquitination-mediated internalization and degradation of the astroglial glutamate transporter, GLT-1. Neurochem Int 53: 296-308. doi: 10.1016/j.neuint.2008.07.010
|
| [105] |
Martinez-Villarreal J, Garcia Tardon N, Ibanez I, et al. (2012) Cell surface turnover of the glutamate transporter GLT-1 is mediated by ubiquitination/deubiquitination. Glia 60: 1356-1365. doi: 10.1002/glia.22354
|
| [106] |
Sheldon AL, Gonzalez MI, Robinson MB (2006) A carboxyl-terminal determinant of the neuronal glutamate transporter, EAAC1, is required for platelet-derived growth factor-dependent trafficking. J Biol Chem 281: 4876-4886. doi: 10.1074/jbc.M504983200
|
| [107] |
Garcia-Tardon N, Gonzalez-Gonzalez IM, Martinez-Villarreal J, et al. (2012) Protein kinase C (PKC)-promoted endocytosis of glutamate transporter GLT-1 requires ubiquitin ligase Nedd4-2-dependent ubiquitination but not phosphorylation. J Biol Chem 287: 19177-19187. doi: 10.1074/jbc.M112.355909
|
| [108] |
A DA, Soragna A, Di Cairano E, et al. (2010) The Surface Density of the Glutamate Transporter EAAC1 is Controlled by Interactions with PDZK1 and AP2 Adaptor Complexes. Traffic 11: 1455-1470. doi: 10.1111/j.1600-0854.2010.01110.x
|
| [109] |
Traub LM (2009) Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat Rev Mol Cell Biol 10: 583-596. doi: 10.1038/nrm2751
|
| [110] |
Sato K, Otsu W, Otsuka Y, et al. (2013) Modulatory roles of NHERF1 and NHERF2 in cell surface expression of the glutamate transporter GLAST. Biochem Biophys Res Commun 430: 839-845. doi: 10.1016/j.bbrc.2012.11.059
|
| [111] | Shouffani A, Kanner BI (1990) Cholesterol is required for the reconstruction of the sodium- and chloride-coupled, gamma-aminobutyric acid transporter from rat brain. J Biol Chem 265: 6002-6008. |
| [112] |
Butchbach ME, Guo H, Lin CL (2003) Methyl-beta-cyclodextrin but not retinoic acid reduces EAAT3-mediated glutamate uptake and increases GTRAP3-18 expression. J Neurochem 84: 891-894. doi: 10.1046/j.1471-4159.2003.01588.x
|
| [113] |
Simons K, Gerl MJ (2010) Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol 11: 688-699. doi: 10.1038/nrm2977
|
| [114] |
Butchbach ME, Tian G, Guo H, et al. (2004) Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem 279: 34388-34396. doi: 10.1074/jbc.M403938200
|
| [115] |
Zschocke J, Bayatti N, Behl C (2005) Caveolin and GLT-1 gene expression is reciprocally regulated in primary astrocytes: association of GLT-1 with non-caveolar lipid rafts. Glia 49: 275-287. doi: 10.1002/glia.20116
|
| [116] |
Gonzalez MI, Krizman-Genda E, Robinson MB (2007) Caveolin-1 regulates the delivery and endocytosis of the glutamate transporter, excitatory amino acid carrier 1. J Biol Chem 282: 29855-29865. doi: 10.1074/jbc.M704738200
|
| [117] | Ledesma MD, Dotti CG (2005) The conflicting role of brain cholesterol in Alzheimer's disease: lessons from the brain plasminogen system. Biochem Soc Symp: 129-138. |
| [118] |
Tian G, Kong Q, Lai L, et al. (2010) Increased expression of cholesterol 24S-hydroxylase results in disruption of glial glutamate transporter EAAT2 association with lipid rafts: a potential role in Alzheimer's disease. J Neurochem 113: 978-989. doi: 10.1111/j.1471-4159.2010.06661.x
|
| [119] |
Arriza JL, Eliasof S, Kavanaugh MP, et al. (1997) Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci U S A 94: 4155-4160. doi: 10.1073/pnas.94.8.4155
|
| [120] | Arriza JL, Fairman WA, Wadiche JI, et al. (1994) Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14: 5559-5569. |
| [121] | Bridges RJ, Stanley MS, Anderson MW, et al. (1991) Conformationally defined neurotransmitter analogues. Selective inhibition of glutamate uptake by one pyrrolidine-2,4-dicarboxylate diastereomer. J Med Chem 34: 717-725. |
| [122] |
Griffiths R, Dunlop J, Gorman A, et al. (1994) L-Trans-Pyrrolidine-2,4-Dicarboxylate and Cis-1-Aminocyclobutane-1,3-Dicarboxylate Behave as Transportable, Competitive Inhibitors of the High-Affinity Glutamate Transporters. Biochem Pharmacol 47: 267-274. doi: 10.1016/0006-2952(94)90016-7
|
| [123] | Vandenberg RJ, Mitrovic AD, Chebib M, et al. (1997) Contrasting modes of action of methylglutamate derivatives on the excitatory amino acid transporters, EAAT1 and EAAT2. Mol Pharmacol 51: 809-815. |
| [124] |
Huang S, Ryan RM, Vandenberg RJ (2009) The role of cation binding in determining substrate selectivity of glutamate transporters. J Biol Chem 284: 4510-4515. doi: 10.1074/jbc.M808495200
|
| [125] |
Eliasof S, McIlvain HB, Petroski RE, et al. (2001) Pharmacological characterization of threo-3-methylglutamic acid with excitatory amino acid transporters in native and recombinant systems. J Neurochem 77: 550-557. doi: 10.1046/j.1471-4159.2001.00253.x
|
| [126] |
Kanai Y, Hediger MA (1992) Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 360: 467-471. doi: 10.1038/360467a0
|
| [127] |
Rauen T, Jeserich G, Danbolt NC, et al. (1992) Comparative analysis of sodium-dependent L-glutamate transport of synaptosomal and astroglial membrane vesicles from mouse cortex. FEBS Lett 312: 15-20. doi: 10.1016/0014-5793(92)81401-7
|
| [128] | Zerangue N, Kavanaugh MP (1996) Interaction of L-cysteine with a human excitatory amino acid transporter. J Physiol 493 ( Pt 2): 419-423. |
| [129] |
Roberts PJ, Watkins JC (1975) Structural requirements for the inhibition for L-glutamate uptake by glia and nerve endings. Brain Res 85: 120-125. doi: 10.1016/0006-8993(75)91016-1
|
| [130] | Wilson DF, Pastuszko A (1986) Transport of Cysteate by Synaptosomes Isolated from Rat-Brain - Evidence That It Utilizes the Same Transporter as Aspartate, Glutamate, and Cysteine Sulfinate. J Neurochem 47: 1091-1097. |
| [131] |
Vandenberg RJ, Mitrovic AD, Johnston GAR (1998) Serine-O-sulphate transport by the human glutamate transporter, EAAT2. Br J Pharmacol 123: 1593-1600. doi: 10.1038/sj.bjp.0701776
|
| [132] |
Bender AS, Woodbury DM, White HS (1989) Beta-Dl-Methylene-Aspartate, an Inhibitor of Aspartate-Aminotransferase, Potently Inhibits L-Glutamate Uptake into Astrocytes. Neurochem Res 14: 641-646. doi: 10.1007/BF00964873
|
| [133] |
Mitrovic AD, Amara SG, Johnston GA, et al. (1998) Identification of functional domains of the human glutamate transporters EAAT1 and EAAT2. J Biol Chem 273: 14698-14706. doi: 10.1074/jbc.273.24.14698
|
| [134] |
Vandenberg RJ, Mitrovic AD, Johnston GA (1998) Serine-O-sulphate transport by the human glutamate transporter, EAAT2. Br J Pharmacol 123: 1593-1600. doi: 10.1038/sj.bjp.0701776
|
| [135] | Campiani G, De Angelis M, Armaroli S, et al. (2001) A rational approach to the design of selective substrates and potent nontransportable inhibitors of the excitatory amino acid transporter EAAC1 (EAAT3). New glutamate and aspartate analogues as potential neuroprotective agents. J Med Chem 44: 2507-2510. |
| [136] |
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65: 1-105. doi: 10.1016/S0301-0082(00)00067-8
|
| [137] |
Wang GJ, Chung HJ, Schnuer J, et al. (1998) Dihydrokainate-sensitive neuronal glutamate transport is required for protection of rat cortical neurons in culture against synaptically released glutamate. Eur J Neurosci 10: 2523-2531. doi: 10.1046/j.1460-9568.1998.00256.x
|
| [138] | Shimamoto K, Lebrun B, Yasuda-Kamatani Y, et al. (1998) DL-threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol 53: 195-201. |
| [139] |
Boudker O, Verdon G (2010) Structural perspectives on secondary active transporters. Trends Pharmacol Sci 31: 418-426. doi: 10.1016/j.tips.2010.06.004
|
| [140] | Shigeri Y, Shimamoto K, Yasuda-Kamatani Y, et al. (2001) Effects of threo-beta-hydroxyaspartate derivatives on excitatory amino acid transporters (EAAT4 and EAAT5). J Neurochem 79: 297-302. |
| [141] |
Shimamoto K, Shigeri Y, Yasuda-Kamatani Y, et al. (2000) Syntheses of optically pure beta-hydroxyaspartate derivatives as glutamate transporter blockers. Bioorg Med Chem Lett 10: 2407-2410. doi: 10.1016/S0960-894X(00)00487-X
|
| [142] |
Lebrun B, Sakaitani M, Shimamoto K, et al. (1997) New beta-hydroxyaspartate derivatives are competitive blockers for the bovine glutamate/aspartate transporter. J Biol Chem 272: 20336-20339. doi: 10.1074/jbc.272.33.20336
|
| [143] |
Shimamoto K, Sakai R, Takaoka K, et al. (2004) Characterization of novel L-threo-beta-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol 65: 1008-1015. doi: 10.1124/mol.65.4.1008
|
| [144] | Shimamoto K, Otsubo Y, Shigeri Y, et al. (2007) Characterization of the tritium-labeled analog of L-threo-beta-benzyloxyaspartate binding to glutamate transporters. Mol Pharmacol 71: 294-302. |
| [145] |
Martinov V, Dehnes Y, Holmseth S, et al. (2014) A novel glutamate transporter blocker, LL-TBOA, attenuates ischaemic injury in the isolated, perfused rat heart despite low transporter levels. Eur J Cardiothorac Surg 45: 710-716. doi: 10.1093/ejcts/ezt487
|
| [146] | Dunlop J, Eliasof S, Stack G, et al. (2003) WAY-855 (3-amino-tricyclo[2.2.1.02.6]heptane-1,3-dicarboxylic acid): a novel, EAAT2-preferring, nonsubstrate inhibitor of high-affinity glutamate uptake. Br J Pharmacol 140: 839-846. |
| [147] |
Dunlop J, McIlvain HB, Carrick TA, et al. (2005) Characterization of novel aryl-ether, biaryl, and fluorene aspartic acid and diaminopropionic acid analogs as potent inhibitors of the high-affinity glutamate transporter EAAT2. Mol Pharmacol 68: 974-982. doi: 10.1124/mol.105.012005
|
| [148] |
Campiani G, Fattorusso C, De Angelis M, et al. (2003) Neuronal high-affinity sodium-dependent glutamate transporters (EAATs): targets for the development of novel therapeutics against neurodegenerative diseases. Curr Pharm Des 9: 599-625. doi: 10.2174/1381612033391261
|
| [149] | Funicello M, Conti P, De Amici M, et al. (2004) Dissociation of [3H]L-glutamate uptake from L-glutamate-induced [3H]D-aspartate release by 3-hydroxy-4,5,6,6a-tetrahydro-3aH-pyrrolo[3,4-d]isoxazole-4-carboxylic acid and 3-hydroxy-4,5,6,6a-tetrahydro-3aH-pyrrolo[3,4-d]isoxazole-6-carboxylic acid, two conformationally constrained aspartate and glutamate analogs. Mol Pharmacol 66: 522-529. |
| [150] |
Callender R, Gameiro A, Pinto A, et al. (2012) Mechanism of inhibition of the glutamate transporter EAAC1 by the conformationally constrained glutamate analogue (+)-HIP-B. Biochemistry 51: 5486-5495. doi: 10.1021/bi3006048
|
| [151] |
Erichsen MN, Huynh TH, Abrahamsen B, et al. (2010) Structure-activity relationship study of first selective inhibitor of excitatory amino acid transporter subtype 1: 2-Amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (UCPH-101). J Med Chem 53: 7180-7191. doi: 10.1021/jm1009154
|
| [152] |
Huynh THV, Shim I, Bohr H, et al. (2012) Structure-Activity Relationship Study of Selective Excitatory Amino Acid Transporter Subtype 1 (EAAT1) Inhibitor 2-Amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (UCPH-101) and Absolute Configurational Assignment Using Infrared and Vibrational Circular Dichroism Spectroscopy in Combination with ab Initio Hartree-Fock Calculations. J Med Chem 55: 5403-5412. doi: 10.1021/jm300345z
|
| [153] |
Abrahamsen B, Schneider N, Erichsen MN, et al. (2013) Allosteric Modulation of an Excitatory Amino Acid Transporter: The Subtype-Selective Inhibitor UCPH-101 Exerts Sustained Inhibition of EAAT1 through an Intramonomeric Site in the Trimerization Domain. J Neurosci 33: 1068-1087. doi: 10.1523/JNEUROSCI.3396-12.2013
|
| [154] |
Rothstein JD, Patel S, Regan MR, et al. (2005) Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433: 73-77. doi: 10.1038/nature03180
|
| [155] |
Fontana AC, de Oliveira Beleboni R, Wojewodzic MW, et al. (2007) Enhancing glutamate transport: mechanism of action of Parawixin1, a neuroprotective compound from Parawixia bistriata spider venom. Mol Pharmacol 72: 1228-1237. doi: 10.1124/mol.107.037127
|
| [156] |
Fontana ACK, Guizzo R, Beleboni RD, et al. (2003) Purification of a neuroprotective component of Parawixia bistriata spider venom that enhances glutamate uptake. Br J Pharmacol 139: 1297-1309. doi: 10.1038/sj.bjp.0705352
|
| [157] |
Xing XC, Chang LC, Kong QM, et al. (2011) Structure-activity relationship study of pyridazine derivatives as glutamate transporter EAAT2 activators. Bioorg Med Chem Lett 21: 5774-5777. doi: 10.1016/j.bmcl.2011.08.009
|
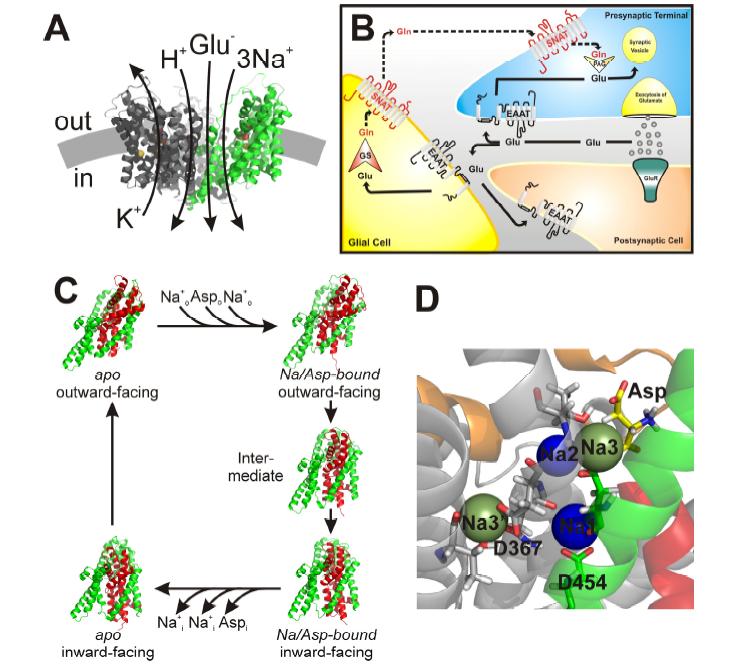

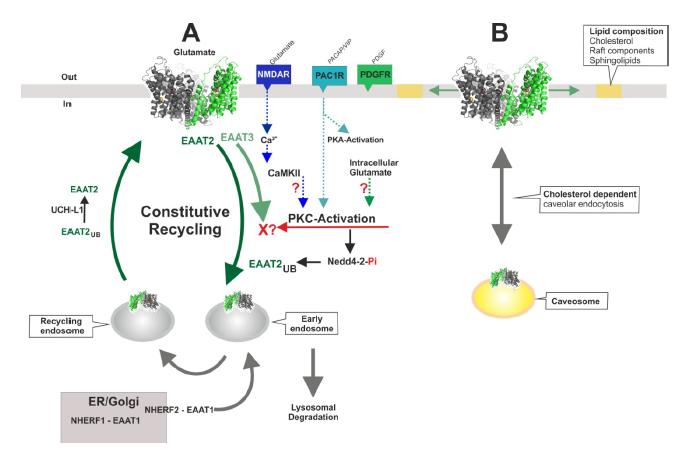
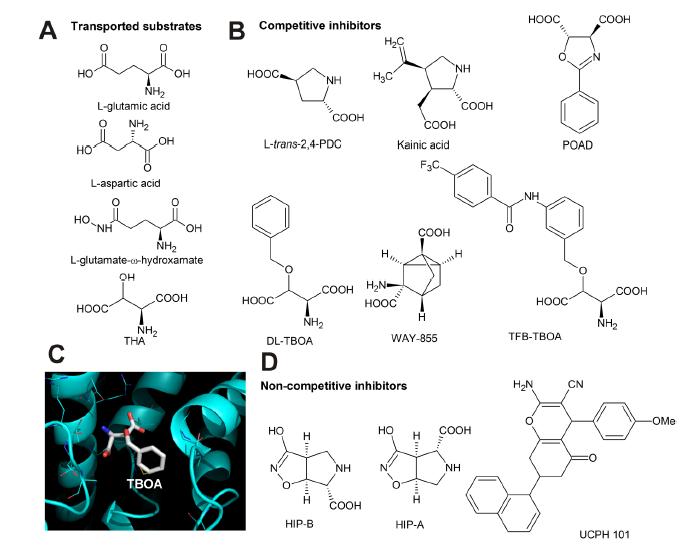
Figures(4)

Thomas Rauen, Rose Tanui, Christof Grewer. Structural and functional dynamics of Excitatory Amino Acid Transporters (EAAT)[J]. AIMS Molecular Science, 2014, 1(3): 99-125. doi: 10.3934/molsci.2014.3.99







 DownLoad:
DownLoad: