Citation: Yuri Pevzner, Daniel N. Santiago, Jacqueline L. von Salm, Rainer S. Metcalf, Kenyon G. Daniel, Laurent Calcul, H. Lee Woodcock, Bill J. Baker, Wayne C. Guida, Wesley H. Brooks. Virtual target screening to rapidly identify potential protein targets of natural products in drug discovery[J]. AIMS Molecular Science, 2014, 1(2): 81-98. doi: 10.3934/molsci.2014.2.81

| [1] | The Florida Center of Excellence in Drug Discovery and Innovation (CDDI) is described at: http://www.research.usf.edu/cddi/drugdiscovery/resources.asp. |
| [2] |
Huang YM, Amsler AO, McClintock JB, et al. (2007) Patterns of gammarid amphipod abundance and species composition associated with dominant subtidal macroalgae along the western Antarctic Peninsula. Polar Biol 30: 1417-1430. doi: 10.1007/s00300-007-0303-1

|
| [3] |
Santiago DN, Pevzner Y, Durand AA, et al. (2012) Virtual Target Screening: Validation Using Kinase Inhibitors. J Chem Inf Model 52: 2192-2203. doi: 10.1021/ci300073m
|
| [4] |
Irwin JJ, Sterling T, Mysinger MM, et al. (2012) ZINC: A Free Tool to Discover Chemistry for Biology. J Chem Info Model 52:1757-1768. doi: 10.1021/ci3001277
|
| [5] |
Gerwick WH, Moore BS (2012) Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem Biol 19:85-98. doi: 10.1016/j.chembiol.2011.12.014
|
| [6] |
Swinney DC, Anthony J (2011) How were new medicines discovered? Nat Rev Drug Discov 10: 507-519. doi: 10.1038/nrd3480
|
| [7] |
Newman DJ, Cragg GM (2012) Natural Products as Sources of New Drugs over the 30 Years from 1981 to 2010. J Nat Prod 75: 311-335. doi: 10.1021/np200906s
|
| [8] |
Mishra BB, Tiwari VK (2011) Natural products: An evolving role in future drug discovery. Eur J Med Chem 46: 4769-4807. doi: 10.1016/j.ejmech.2011.07.057
|
| [9] |
Bachmann BO, Van Lanen SG, Baltz RH (2014) Microbial genome mining for accelerated natural products discovery: is a renaissance in the making? J Indust Microbiol Biotech 41: 175-184. doi: 10.1007/s10295-013-1389-9
|
| [10] |
Williams P, Sorribasa A, Howes MR (2011) Natural products as a source of Alzheimer's drug leads. Nat Prod Rep 28: 48-77. doi: 10.1039/C0NP00027B
|
| [11] |
Kuete V, Alibert-Franco S, Eyong KO, et al. (2011) Antibacterial activity of some natural products against bacteria expressing a multidrug-resistant phenotype. Int J Antimicrob Agents 37: 156-161. doi: 10.1016/j.ijantimicag.2010.10.020
|
| [12] |
Cragg GM, Newman DJ (2013) Natural products: A continuing source of novel drug leads. Biochim Biophys Acta 1830: 3670-3695. doi: 10.1016/j.bbagen.2013.02.008
|
| [13] |
Dayan FE, Owens DK, Duke SO (2012) Rationale for a natural products approach to herbicide discovery. Pest Mgmt Sci 68: 519-528. doi: 10.1002/ps.2332
|
| [14] |
Bugni TS, Harper MK, McCulloch MWB, et al. (2008) Fractionated marine invertebrate extract libraries for drug discovery. Molecules 13: 1372-1383. doi: 10.3390/molecules13061372
|
| [15] |
Molinski TF (2010) NMR of natural products at the “nanomole-scale”. Nat Prod Rep 27: 321-329. doi: 10.1039/b920545b
|
| [16] |
Nicolaou KC, Snyder SA (2005) Chasing molecules that were never there: misassigned natural products and the role of chemical synthesis in modern structure elucidation. Angew. Chem Int Ed Engl 44: 1012-1044. doi: 10.1002/anie.200460864
|
| [17] |
Penn K, Jenkins C, Nett M, et al. (2009) Genomic islands link secondary metabolism to functional adaptation in marine Actinobacteria. ISME J 3: 1193-1203. doi: 10.1038/ismej.2009.58
|
| [18] |
Lipinski CA, Lombardo F, Dominy BW, et al. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46: 3-26. doi: 10.1016/S0169-409X(00)00129-0
|
| [19] |
Ursu O, Rayan A, Goldblum A, et al. (2011) Understanding drug-likeness. WIREs: Comp Mol Sci 1:760-781. doi: 10.1002/wcms.52
|
| [20] |
Bickerton GR, Paoliuni GV, Besnard J, et al. (2012) Quantifying the chemical beauty of drugs. Nat Chem 4: 90-98. doi: 10.1038/nchem.1243
|
| [21] | Matter H, Sotriffer C (2011) Applications and success stories in virtual screening. In: Virtual screening: principles, challenges, and practical guidelines. Wiley-VCH Verlag GmbH & Co. Ch. 12: 319-358. |
| [22] |
Vangrevelinghe E, Zimmermann K, Schoepfer J, et al. (2003) Discovery of a Potent and Selective Protein Kinase CK2 Inhibitor by High-Throughput Docking. J Med Chem 46: 2656-2662. doi: 10.1021/jm030827e
|
| [23] | Ewing TA, Makino S, Skillman AG, et al. (2001) DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comp Mol Des 15: 411-428. |
| [24] | Friesner RA, Banks JL, Murphy RB, et al. (2004) Glide: New approach for rapid, accurate docking and scoring. 1. Method and Assessment of Docking Accuracy. J Med Chem 47: 1739-1749. |
| [25] |
26. Brooks WH, McCloskey DE, Daniel KE, et al. (2007) In Silico Chemical Library Screening and Experimental Validation of a Novel 9-Aminoacridine Based Lead-Inhibitor of Human S-Adenosylmethionine Decarboxylase. J Chem Info Model 47: 1897-1905. doi: 10.1021/ci700005t
|
| [26] |
27. Tolbert WD, Ekstrom JL, Mathews II, et al. (2001) The structural basis for substrate specificity and inhibition of human S-adenosylmethionine decarboxylase. Biochemistry 40: 9484-9494. doi: 10.1021/bi010735w
|
| [27] |
28. Berman HM, Westbrook J, Feng Z, et al. (2000) The Protein Data Bank. Nucl Acids Res 28: 235-242. doi: 10.1093/nar/28.1.235
|
| [28] |
29. Hui-Fang L, Qing S, Jian Z, et al. (2010) Evaluation of various inverse docking schemes in multiple targets identification. J Mol Graph Model 29: 326-330. doi: 10.1016/j.jmgm.2010.09.004
|
| [29] |
30. Grinter SZ, Liang Y, Huang SY, et al. (2011) An inverse docking approach for identifying new potential anti-cancer targets. J Mol Graph Model 795-799. doi: 10.1016/j.jmgm.2011.01.002
|
| [30] |
31. Do QT, Lamy C, Renimel I, et al. (2007) Reverse Pharmacognosy: Identifying Biological Properties for Plants by Means of their Molecule Constituents: Application to Meranzin. Planta Med 73: 1235-1240. doi: 10.1055/s-2007-990216
|
| [31] | 32. Li YY, An J, Jones SJM (2006) A Large-Scale Computational Approach to Drug Repositioning. Genome Info 17: 239-247. |
| [32] |
37. Miller BT, Singh RP, Klauda JB, et al. (2008) CHARMMing: a new, flexible web portal for CHARMM. J Chem Info Model 48: 1920-1929. doi: 10.1021/ci800133b
|
| [33] |
38. Wang Y, Xiao J, Suzek TO, et al. (2012) PubChem's BioAssay Database. Nucl Acids Res 40: D400-12. doi: 10.1093/nar/gkr1132
|
| [34] | 39. Bolton E, Wang Y, Thiessen PA, et al. (2008) PubChem: Integrated Platform of Small Molecules and Biological Activities. Chapter 12 in Annual Reports in Computational Chemistry, Volume 4, ACS, Washington, DC. |
| [35] |
42. Imhoff JF, Labes A, Wiese J (2011) Bio-mining the microbial treasures of the ocean: New natural products. Biotech Adv 29: 468-482. doi: 10.1016/j.biotechadv.2011.03.001
|
| [36] |
43. Liu X, Ashforth E, Ren B, et al. (2010) Bioprospecting microbial natural product libraries from the marine environment for drug discovery. J Antibiotics 63: 415-422. doi: 10.1038/ja.2010.56
|
| [37] |
44. Bauer RA, Wurst JM, Tan DS (2010) Expanding the range of “druggable” targets with natural product-based libraries: an academic perspective. Curr Op Chem Biol 14: 308-314. doi: 10.1016/j.cbpa.2010.02.001
|
| [38] |
45. Mayer AMS, Glaser KB, Cuevas C, et al. (2010) The odyssey of marine pharmaceuticals: a current pipeline perspective. Trends in Pharm Sci 31: 255-265. doi: 10.1016/j.tips.2010.02.005
|
| [39] | 46. Bennani YL (2011) Drug discovery in the next decade: innovation needed ASAP. Drug Disc Today 16: |
| [40] |
47. Keiser MJ, Setola V, Irwin JJ, et al. (2009) Predicting new molecular targets for known drugs. Nature 462: 175-182. doi: 10.1038/nature08506
|
| [41] |
48. Reker D, Rodrigues T, Schneider P, et al. (2014) Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc Natl Acad Sci USA 111: 4067-4072. doi: 10.1073/pnas.1320001111
|
| [42] |
49. Liu X, Vogt I, Hague T, et al. (2013) HitPick: A web server for hit identification and target prediction of chemical screenings. Bioinformatics 29: 1910-1912. doi: 10.1093/bioinformatics/btt303
|
| [43] |
50. Gong J, Cai C, Liu X, et al. (2013) ChemMapper: A versatile web server for exploring pharmacology and chemical structure association based on molecular 3D similarity method. Bioinformatics 29: 1827-1829. doi: 10.1093/bioinformatics/btt270
|
| [44] |
51. Iorio F, Bosotti R, Scachen E, et al. (2010) Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc Nat Acad Sic USA 107: 14621-14626. doi: 10.1073/pnas.1000138107
|
| [45] |
52. Zhu X, Kim JL, Newcomb JR, et al. (1999) Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Struct Fold Des 7: 651-661. doi: 10.1016/S0969-2126(99)80086-0
|
| [46] |
53. Goto M, Miyahara I, Hirotsu K, et al. (2005) Structural determinants for branched-chain aminotransferase isozyme-specific inhibition by the anticonvulsant drug gabapentin. J Biol Chem 280: 37237256. doi: 10.1074/jbc.M506486200
|
| [47] |
55. Halgren T (2009) Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model 49:377-389. doi: 10.1021/ci800324m
|
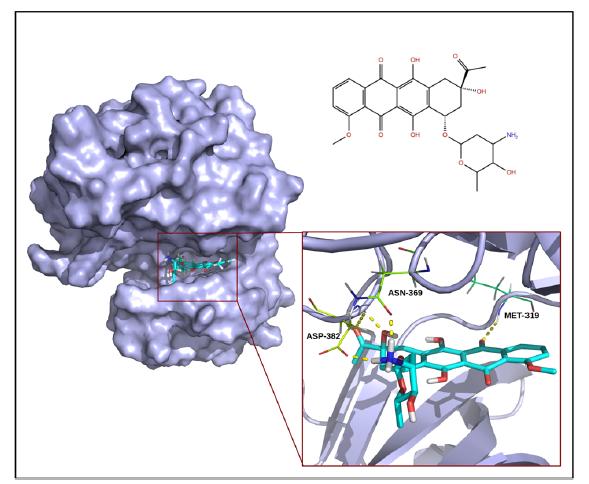
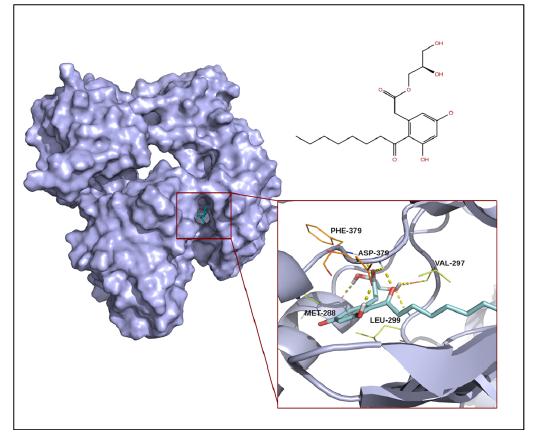
Figures(2) / Tables(2)

Yuri Pevzner, Daniel N. Santiago, Jacqueline L. von Salm, Rainer S. Metcalf, Kenyon G. Daniel, Laurent Calcul, H. Lee Woodcock, Bill J. Baker, Wayne C. Guida, Wesley H. Brooks. Virtual target screening to rapidly identify potential protein targets of natural products in drug discovery[J]. AIMS Molecular Science, 2014, 1(2): 81-98. doi: 10.3934/molsci.2014.2.81







 DownLoad:
DownLoad: