Citation: Christopher Chong, Andre Foehr, Efstathios G. Charalampidis, Panayotis G. Kevrekidis, Chiara Daraio. Breathers and other time-periodic solutions in an array of cantilevers decorated with magnets[J]. Mathematics in Engineering, 2019, 1(3): 489-507. doi: 10.3934/mine.2019.3.489

| [1] | Gallavotti G (2008) The Fermi–Pasta–Ulam Problem: A Status Report. Heidelberg: Springer-Verlag. |
| [2] |
Kevrekidis PG (2011) Non-linear waves in lattices: Past, present, future. IMA J Appl Math 76: 389–423. doi: 10.1093/imamat/hxr015

|
| [3] |
Lederer F, Stegeman GI, Christodoulides DN, et al. (2008) Discrete solitons in optics. Phys Rep 463: 1–126. doi: 10.1016/j.physrep.2008.04.004
|
| [4] |
Binder P, Abraimov D, Ustinov AV, et al. (2000) Observation of breathers in Josephson ladders. Phys Rev Lett 84: 745–748. doi: 10.1103/PhysRevLett.84.745
|
| [5] |
Trías E, Mazo JJ, Orlando TP (2000) Discrete breathers in nonlinear lattices: Experimental detection in a Josephson array. Phys Rev Lett 84: 741–744. doi: 10.1103/PhysRevLett.84.741
|
| [6] | English LQ, Sato M, Sievers AJ (2003) Modulational instability of nonlinear spin waves in easy-axis antiferromagnetic chains. II. Influence of sample shape on intrinsic localized modes and dynamic spin defects. Phys Rev B 67: 024403. |
| [7] |
Schwarz UT, English LQ, Sievers AJ (1999) Experimental generation and observation of intrinsic localized spin wave modes in an antiferromagnet. Phys Rev Lett 83: 223–226. doi: 10.1103/PhysRevLett.83.223
|
| [8] |
Swanson BI, Brozik JA, Love SP, et al. (1999) Observation of intrinsically localized modes in a discrete low-dimensional material. Phys Rev Lett 82: 3288–3291. doi: 10.1103/PhysRevLett.82.3288
|
| [9] |
Peyrard M (2004) Nonlinear dynamics and statistical physics of DNA. Nonlinearity 17: R1–R40. doi: 10.1088/0951-7715/17/2/R01
|
| [10] |
Morsch O, Oberthaler M (2006) Dynamics of Bose-Einstein condensates in optical lattices. Rev Mod Phys 78: 179–215. doi: 10.1103/RevModPhys.78.179
|
| [11] | Nesterenko VF (2001) Dynamics of Heterogeneous Materials. New York: Springer-Verlag, USA. |
| [12] | Chong C, Kevrekidis P (2018) Coherent Structures in Granular Crystals: From Experiment and Modelling to Computation and Mathematical Analysis. Heidelberg: Springer. |
| [13] | Starosvetsky Y, Jayaprakash KR, Hasan MA, et al. (2017) Dynamics and Acoustics of Ordered Granular Media. Singapore: World Scientific. |
| [14] |
Rosas A, Lindenberg K (2018) Pulse propagation in granular chains. Phys Rep 735: 1–37. doi: 10.1016/j.physrep.2018.02.001
|
| [15] |
Chong C, Porter MA, Kevrekidis PG, et al. (2017) Nonlinear coherent structures in granular crystals. J Phys Condens Matter 29: 413002. doi: 10.1088/1361-648X/aa7e36
|
| [16] |
Sen S, Hong J, Bang J, et al. (2008) Solitary waves in the granular chain. Phys Rep 462: 21–66. doi: 10.1016/j.physrep.2007.10.007
|
| [17] |
Bonanomi L, Theocharis G, Daraio C (2015) Wave propagation in granular chains with local resonances. Phys Rev E 91: 033208. doi: 10.1103/PhysRevE.91.033208
|
| [18] |
Lydon J, Serra-Garcia M, Daraio C (2014) Local to extended transitions of resonant defect modes. Phys Rev Lett 113: 185503. doi: 10.1103/PhysRevLett.113.185503
|
| [19] |
James G (2011) Nonlinear waves in Newton's cradle and the discrete p-Schrödinger equation. Math Models Methods Appl Sci 21: 2335–2377. doi: 10.1142/S0218202511005763
|
| [20] |
Kimura M, Hikihara T (2008) Stability change of intrinsic localized mode in finite nonlinear coupled oscillators. Phys Lett A 372: 4592–4595. doi: 10.1016/j.physleta.2008.04.054
|
| [21] |
Sato M, Hubbard BE, Sievers AJ (2006) Colloquium: Nonlinear energy localization and its manipulation in micromechanical oscillator arrays. Rev Mod Phys 78: 137–157. doi: 10.1103/RevModPhys.78.137
|
| [22] | Erturk A, Inman DJ (2011) Piezoelectric Energy Harvesting. Hoboken: Wiley & Sons. |
| [23] |
Giannoulis J, Mielke A (2004) The nonlinear Schrödinger equation as a macroscopic limit for an oscillator chain with cubic nonlinearities. Nonlinearity 17: 551–565. doi: 10.1088/0951-7715/17/2/011
|
| [24] |
Theocharis G, Kavousanakis M, Kevrekidis PG, et al. (2009) Localized breathing modes in granular crystals with defects. Phys Rev E 80: 066601. doi: 10.1103/PhysRevE.80.066601
|
| [25] |
Boechler N, Theocharis G, Daraio C (2011) Bifurcation based acoustic switching and rectification. Nat Mater 10: 665–668. doi: 10.1038/nmat3072
|
| [26] |
Huang G, Shi ZP, Xu Z (1993) Asymmetric intrinsic localized modes in a homogeneous lattice with cubic and quartic anharmonicity. Phys Rev B 47: 14561–14564. doi: 10.1103/PhysRevB.47.14561
|
| [27] |
Nadkarni N, Arrieta AF, Chong C, et al. (2016) Unidirectional transition waves in bistable lattices. Phys Rev Lett 116: 244501. doi: 10.1103/PhysRevLett.116.244501
|
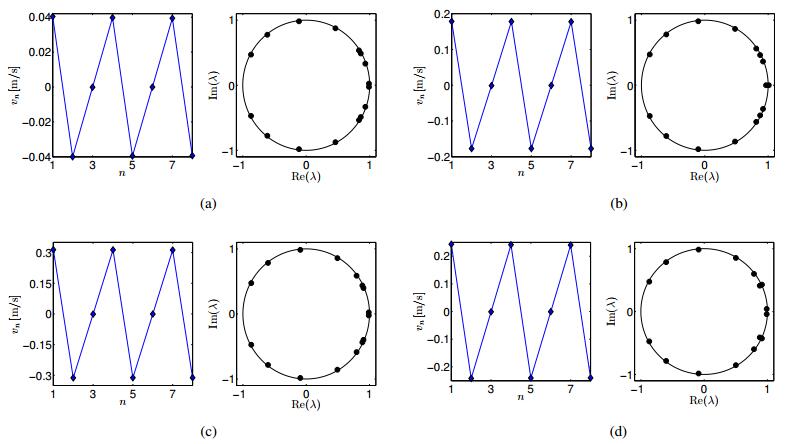
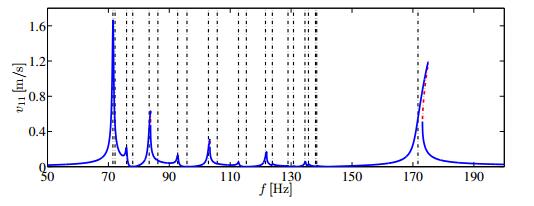
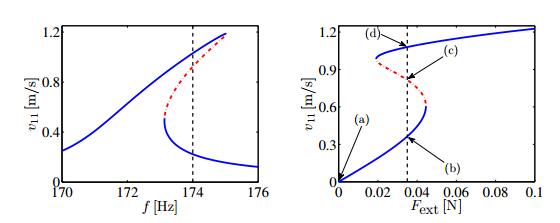
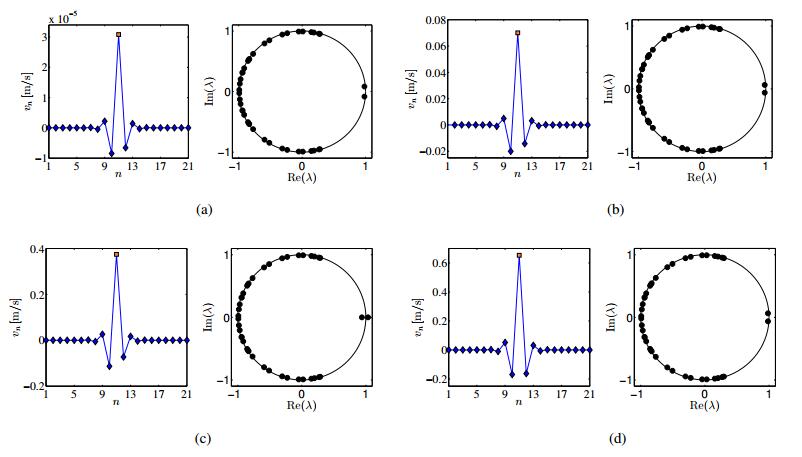
Figures(14) / Tables(2)

Christopher Chong, Andre Foehr, Efstathios G. Charalampidis, Panayotis G. Kevrekidis, Chiara Daraio. Breathers and other time-periodic solutions in an array of cantilevers decorated with magnets[J]. Mathematics in Engineering, 2019, 1(3): 489-507. doi: 10.3934/mine.2019.3.489







 DownLoad:
DownLoad: