Citation: Li Bin, Muhammad Shahzad, Qi Bing, Muhammad Ahsan, Muhammad U Shoukat, Hafiz MA Khan, Nabeel AM Fahal. The probabilistic load flow analysis by considering uncertainty with correlated loads and photovoltaic generation using Copula theory[J]. AIMS Energy, 2018, 6(3): 414-435. doi: 10.3934/energy.2018.3.414

| [1] |
Bechrakis DA, Sparis PD (2004) Correlation of wind speed between neighboring measuring stations. IEEE T Energy Conver 19: 400–406. doi: 10.1109/TEC.2004.827040

|
| [2] | Villanueva D, Feijóo A, Pazos JL (2010) Correlation between power generated by wind turbines from different locations. Available from: http://proceedings.ewea.org/ewec2010/allfiles2/20_EWEC2010presentation.pdf. |
| [3] | Jong MD, Papaefthymiou G, Palensky P (2017) A framework for incorporation of infeed uncertainty in power system risk-based security assessment. IEEE T Power Syst 33: 613–621. |
| [4] |
Kirschen D, Jayaweera D (2007) Comparison of risk-based and deterministic security assessments. IET Gener Transm Dis 1: 527–533. doi: 10.1049/iet-gtd:20060368
|
| [5] | Zhang P, Lee ST (2004) Probabilistic load flow computation using the method of combined cumulants and Gram-Charlier expansion. IEEE T Power Syst 19: 676–682. |
| [6] |
Nijhuis M, Gibescu M, Cobben S (2017) Gaussian mixture based probabilistic load flow for LV-network planning. IEEE T Power Syst 32: 2878–2886. doi: 10.1109/TPWRS.2016.2628959
|
| [7] |
Valverde G, Saric A, Terzija V (2012) Probabilistic load flow with non-Gaussian correlated random variables using Gaussian mixture models. IET Gener Transm Dis 6: 701–709. doi: 10.1049/iet-gtd.2011.0545
|
| [8] |
Prusty BR, Jena D (2016) Combined cumulant and Gaussian mixture approximation for correlated probabilistic load flow studies: A new approach. Csee J Power Energ Syst 2: 71–78. doi: 10.17775/CSEEJPES.2016.00024
|
| [9] |
Caramia P, Carpinelli G, Varilone P (2010) Point estimate schemes for probabilistic three-phase load flow. Electr Pow Syst Res 80: 168–175. doi: 10.1016/j.epsr.2009.08.020
|
| [10] |
Morales JM, Perez-Ruiz J (2007) Point estimate schemes to solve the probabilistic power flow. IEEE T Power Syst 22: 1594–1601. doi: 10.1109/TPWRS.2007.907515
|
| [11] | Kloubert ML, Rehtanz C (2017) Enhancement to the combination of point estimate method and Gram-Charlier Expansion method for probabilistic load flow computations. PowerTech, IEEE Manchester 2017: 1–6. |
| [12] | Wang Y, Zhang N, Chen Q, et al. (2016) Dependent discrete convolution based probabilistic load flow for the active distribution system. IEEE T Sustain Energ 8: 1000–1009. |
| [13] |
Zhang N, Kang C, Singh C, et al. (2016) Copula based dependent discrete convolution for power system uncertainty analysis. IEEE T Power Syst 31: 5204–5205. doi: 10.1109/TPWRS.2016.2521328
|
| [14] | Cai D, Chen J, Shi D, et al. (2012) Enhancements to the cumulant method for probabilistic load flow studies. Pow Energ Soc Gen Meeting 59: 1–8. |
| [15] |
Aien M, Fotuhi-Firuzabad M, Aminifar F (2012) Probabilistic load flow in correlated uncertain environment using unscented transformation. IEEE T Power Syst 27: 2233–2241. doi: 10.1109/TPWRS.2012.2191804
|
| [16] |
Fang S, Cheng H, Xu G (2016) A modified Nataf transformation-based extended quasi-monte carlo simulation method for solving probabilistic load flow. Electr Mach Pow Syst 44: 1735–1744. doi: 10.1080/15325008.2016.1173130
|
| [17] |
Ren Z, Koh CS (2013) A second-order design sensitivity-assisted Monte Carlo simulation method for reliability evaluation of the electromagnetic devices. J Electr Eng Technol 8: 780–786. doi: 10.5370/JEET.2013.8.4.780
|
| [18] | Tang L, Wen F, Salam MA, et al. (2015) Transmission system planning considering integration of renewable energy resources. Pow Energ Eng Conf 2015: 1–5. |
| [19] | Ayodele TR (2016) Analysis of monte carlo simulation sampling techniques on small signal stability of wind generator-connected power system. J Eng Sci Technol 11: 563–583. |
| [20] |
Prusty BR, Jena D (2017) A critical review on probabilistic load flow studies in uncertainty constrained power systems with photovoltaic generation and a new approach. Renew Sust Energ Rev 69: 1286–1302. doi: 10.1016/j.rser.2016.12.044
|
| [21] |
Morales JM, Baringo L, Conejo AJ, et al. (2010) Probabilistic power flow with correlated wind sources. Let Gener Transm Dis 4: 641–651. doi: 10.1049/iet-gtd.2009.0639
|
| [22] |
Usaola J (2010) Probabilistic load flow with correlated wind power injections. Electr Pow Syst Res 80: 528–536. doi: 10.1016/j.epsr.2009.10.023
|
| [23] |
Gupta N (2016) Probabilistic load flow with detailed wind generator models considering correlated wind generation and correlated loads. Renew Energ 94: 96–105. doi: 10.1016/j.renene.2016.03.030
|
| [24] |
Prusty BR, Jena D (2017) A sensitivity matrix-based temperature-augmented probabilistic load flow study. IEEE T Ind Appl 53: 2506–2516. doi: 10.1109/TIA.2017.2660462
|
| [25] | Kenari MT, Sepasian MS, Nazar MS, et al. (2017) The combined cumulants and Laplace transform method for probabilistic load flow analysis. Let Gener Transm Dis. |
| [26] |
Wu W, Wang K, Li G, et al. (2016) Probabilistic load flow calculation using cumulants and multiple integrals. Let Gener Transm Dis 10: 1703–1709. doi: 10.1049/iet-gtd.2015.1129
|
| [27] | Nelsen RB (2007) An introduction to copulas. Springer Science & Business Media. |
| [28] | Market Analysis and Information System (MAISY). Utility Customer Energy Use & Hourly Load Databases. Available from: http://www.maisy.com/. |
| [29] | National Renewable Energy Laboratory (NREL). Obtain Solar Power data: Western State California. Available from: https://www.nrel.gov/grid/solar-power-data.html. |
| [30] | Macdonald IA (2009) Comparison of sampling techniques on the performance of Monte-Carlo based sensitivity analysis. Eleventh International IBPSA Conference, 992–999. |
| [31] |
Yu H, Chung CY, Wong KP, et al. (2009) Probabilistic load flow evaluation with hybrid latin hypercube sampling and cholesky decomposition. IEEE T Power Syst 24: 661–667. doi: 10.1109/TPWRS.2009.2016589
|
| [32] | University of Washington Electrical Engineering: Power System Test Case Archive (January 2018). Available from: http://www.ee.washington.edu/research/pstca. |
| [33] |
Karaki S, Chedid R, Ramadan R (1999) Probabilistic performance assessment of autonomous solar-wind energy conversion systems. IEEE T Energy Conver 14: 766–772. doi: 10.1109/60.790949
|
| [34] | Reddy JB, Reddy DN (2004) Probabilistic performance assessment of a roof top wind, solar photo voltaic hybrid energy system. Rel Maint S-RAMS 2004: 654–658. |
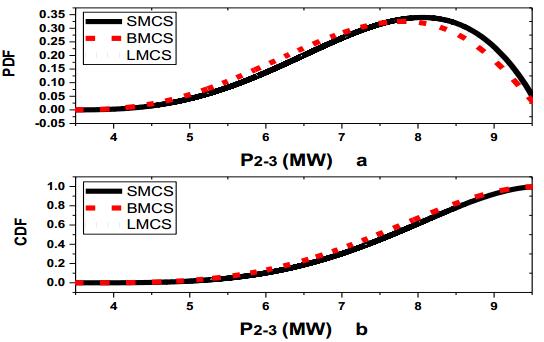
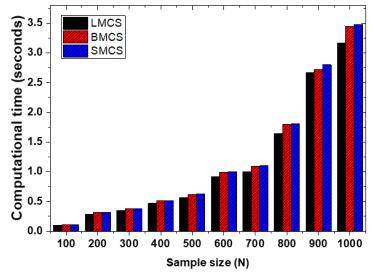
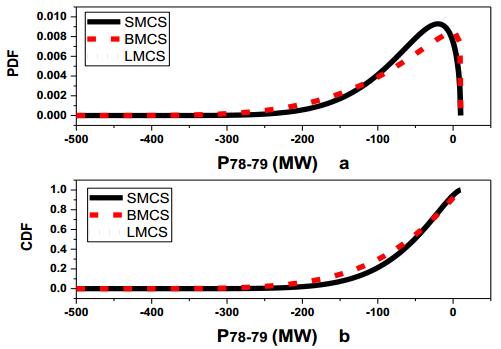
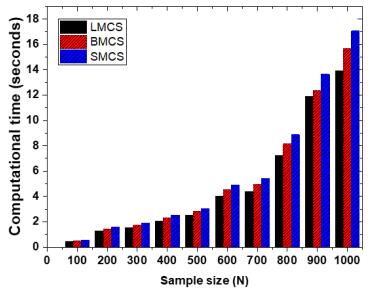
Figures(13) / Tables(3)

Li Bin, Muhammad Shahzad, Qi Bing, Muhammad Ahsan, Muhammad U Shoukat, Hafiz MA Khan, Nabeel AM Fahal. The probabilistic load flow analysis by considering uncertainty with correlated loads and photovoltaic generation using Copula theory[J]. AIMS Energy, 2018, 6(3): 414-435. doi: 10.3934/energy.2018.3.414







 DownLoad:
DownLoad: